Studying gene expression in a cancer patient’s cells can help clinical biologists understand the cancer’s origin and predict the success of different treatments. But cells are complex and contain many layers, so how the biologist conducts measurements affects which data they can obtain. For instance, measuring proteins in a cell could yield different information about the effects of cancer than measuring gene expression or cell morphology.
Where in the cell the information comes from matters. But to capture complete information about the state of the cell, scientists often must conduct many measurements using different techniques and analyze them one at a time. Machine-learning methods can speed up the process, but existing methods lump all the information from each measurement modality together, making it difficult to figure out which data came from which part of the cell.
To overcome this problem, researchers at the Broad Institute of MIT and Harvard and ETH Zurich/Paul Scherrer Institute (PSI) developed an artificial intelligence-driven framework that learns which information about a cell’s state is shared across different measurement modalities and which information is unique to a particular measurement type.
By pinpointing which information came from which cell parts, the approach provides a more holistic view of the cell’s state, making it easier for a biologist to see the complete picture of cellular interactions. This could help scientists understand disease mechanisms and track the progression of cancer, neurodegenerative disorders such as Alzheimer’s, and metabolic diseases like diabetes.
“When we study cells, one measurement is often not sufficient, so scientists develop new technologies to measure different aspects of cells. While we have many ways of looking at a cell, at the end of the day we only have one underlying cell state. By putting the information from all these measurement modalities together in a smarter way, we could have a fuller picture of the state of the cell,” says lead author Xinyi Zhang SM ’22, PhD ’25, a former graduate student in the MIT Department of Electrical Engineering and Computer Science (EECS) and an affiliate of the Eric and Wendy Schmidt Center at the Broad Institute of MIT and Harvard, who is now a group leader at AITHYRA in Vienna, Austria.
Zhang is joined on a paper about the work by G.V. Shivashankar, a professor in the Department of Health Sciences and Technology at ETH Zurich and head of the Laboratory of Multiscale Bioimaging at PSI; and senior author Caroline Uhler, a professor in EECS and the Institute for Data, Systems, and Society (IDSS) at MIT, member of MIT’s Laboratory for Information and Decision Systems (LIDS), and director of the Eric and Wendy Schmidt Center at the Broad Institute. The research appears today in Nature Computational Science.
Manipulating multiple measurements
There are many tools scientists can use to capture information about a cell’s state. For instance, they can measure RNA to see if the cell is growing, or they can measure chromatin morphology to see if the cell is dealing with external physical or chemical signals.
“When scientists perform multimodal analysis, they gather information using multiple measurement modalities and integrate it to better understand the underlying state of the cell. Some information is captured by one modality only, while other information is shared across modalities. To fully understand what is happening inside the cell, it is important to know where the information came from,” says Shivashankar.
Often, for scientists, the only way to sort this out is to conduct multiple individual experiments and compare the results. This slow and cumbersome process limits the amount of information they can gather.
In the new work, the researchers built a machine-learning framework that specifically understands which information overlaps between different modalities, and which information is unique to a particular modality but not captured by others.
“As a user, you can simply input your cell data and it automatically tells you which data are shared and which data are modality-specific,” Zhang says.
To build this framework, the researchers rethought the typical way machine-learning models are designed to capture and interpret multimodal cellular measurements.
Usually these methods, known as autoencoders, have one model for each measurement modality, and each model encodes a separate representation for the data captured by that modality. The representation is a compressed version of the input data that discards any irrelevant details.
The MIT method has a shared representation space where data that overlap between multiple modalities are encoded, as well as separate spaces where unique data from each modality are encoded.
In essence, one can think of it like a Venn diagram of cellular data.
The researchers also used a special, two-step training procedure that helps their model handle the complexity involved in deciding which data are shared across multiple data modalities. After training, the model can identify which data are shared and which are unique when fed cell data it has never seen before.
Distinguishing data
In tests on synthetic datasets, the framework correctly captured known shared and modality-specific information. When they applied their method to real-world single-cell datasets, it comprehensively and automatically distinguished between gene activity captured jointly by two measurement modalities, such as transcriptomics and chromatin accessibility, while also correctly identifying which information came from only one of those modalities.
In addition, the researchers used their method to identify which measurement modality captured a certain protein marker that indicates DNA damage in cancer patients. Knowing where this information came from would help a clinical scientist determine which technique they should use to measure that marker.
“There are too many modalities in a cell and we can’t possibly measure them all, so we need a prediction tool. But then the question is: Which modalities should we measure and which modalities should we predict? Our method can answer that question,” Uhler says.
In the future, the researchers want to enable the model to provide more interpretable information about the state of the cell. They also want to conduct additional experiments to ensure it correctly disentangles cellular information and apply the model to a wider range of clinical questions.
“It is not sufficient to just integrate the information from all these modalities,” Uhler says. “We can learn a lot about the state of a cell if we carefully compare the different modalities to understand how different components of cells regulate each other.”
This research is funded, in part, by the Eric and Wendy Schmidt Center at the Broad Institute, the Swiss National Science Foundation, the U.S. National Institutes of Health, the U.S. Office of Naval Research, AstraZeneca, the MIT-IBM Watson AI Lab, the MIT J-Clinic for Machine Learning and Health, and a Simons Investigator Award.
When Sebastiano Cultrera di Montesano arrived at the Eric and Wendy Schmidt Center at the Broad Institute of MIT and Harvard in 2024, he brought with him a background in mathematics — and a willingness to learn the language of biology.
“He came as mostly a pure math guy,” says Peter Winter, Principal Investigator and Co-Director of the Project Ex Vivo group at the Broad Institute. “He would sit in lab meetings, listening to the biological problems we were dealing with. And then once he learned the language a little bit, he started offering ideas.”
Seb, a postdoctoral fellow at the Schmidt Center, completed his PhD in computational topology and geometry from the Institute of Science and Technology Austria (ISTA) under the supervision of Herbert Edelsbrunner, focusing on uncovering structure in complex data.
Enjoying a walk on Commonwealth Ave
“I was always fascinated by the biomedical sciences,” Seb says. “I thought at some point I would switch from mathematics to biology, but the math kept being intriguing.” After a biotech internship in Paris near the end of his PhD, he began thinking about combining the two. “I remember being captivated by it — developing new mathematical tools for biological questions.”
When he learned about the Schmidt Center, which aims to build a two-way street between machine learning and biology, the fit felt natural. “The Schmidt Center put the emphasis on finding researchers with strong mathematical foundations without requiring deep prior experience in biology, to help uncover biological problems. There aren’t too many places like that.” After visiting the Broad and meeting fellows and faculty, he decided to make the leap across the Atlantic.
Today, his work sits squarely at the intersection of mathematics, AI, and biology. “I thought this would be an incredible place to continue my research.”
Tell us more about your research interests.
“My background is really about structure,” Seb says. “Computational geometry and topology are about asking: what is the shape inside a dataset? How is it organized?”
When he arrived at the Broad, he spent his first weeks talking to researchers across groups.
“It was an adjustment at first,” he admits. “You’re not getting ‘actual work’ done — you’re mostly listening and trying to understand what questions might be interesting.”
One question kept coming back to him:
“When are experiments that biologists run actually predictable? And when do we really need to do them to learn something new?”
He explains it with a simple analogy:
“As humans, there are a lot of experiments in life that we don’t do. If I take a glass of water and throw it on the floor and it breaks, I don’t need to take a mug and throw it on the floor to see whether it will break. We have some understanding of the physical world that lets us decide what to test and what not to test — because we can’t possibly try everything. The floor and broken glass would be a mess!”
He became interested in whether machine learning models could develop a similar intuition for certain biological systems — predicting outcomes in advance and perhaps helping guide which experiments are worth running.
Seb and fellow author and frequent collaborator Davide D’Ascenzo
You recently published a paper in Nature Computational Science. What problem were you trying to solve?
“The task is: if I’m given gene expression data for a cell, can I categorize it into a cell type?” he explains. “Macrophage, T cell, and so on.”
But he noticed something subtle.
“These labels aren’t just a flat list. They’re organized like a tree.”
He offers another analogy:
“If I’m trying to predict something like cat, dog, elephant, or mouse — that’s a flat list. But if I’m trying to predict golden retriever, poodle, or tabby cat, those are at different levels of granularity. Golden retriever is a kind of dog, which is a kind of animal. So the labels have structure.”
Cell types work the same way. There are broad categories like immune cells, more specific ones like T cells, and then even more granular subtypes.
“Most models treat these labels as independent,” Seb says. “If the model predicts something slightly more specific — say CD-positive T cell instead of just T cell — it’s considered completely wrong. But biologically, that’s not entirely reasonable.”
So instead of building a brand-new model, Seb modified the training objective itself.
“I didn’t invent a new architecture,” he says. “I took existing methods and tweaked them so they respect the geometry of the label space.”
The resulting hierarchical cross-entropy loss improved performance across multiple model classes — and, importantly, applies to any hierarchical classification task, not just cell types.
Lorin Crawford, Principal Researcher at Microsoft Research who co-leads Project Ex Vivo, says the elegance stood out immediately. “The idea felt so simple. It was like, ‘Why hasn’t anyone already done this?’ That’s very much Seb — coming up with something that seems obvious in hindsight.”
You collaborated closely with the Project Ex Vivo group. What was that experience like?
Seb first met Lorin while visiting the Broad during his PhD, after reaching out to discuss topology. When he later joined the Schmidt Center, the collaboration deepened naturally.
“The timing was right because Project Ex Vivo’s efforts are similar in spirit to the Schmidt Center – trying to put together researchers of different backgrounds, both biologists and computer scientists,” says Seb.
“It’s been a really wonderful situation,” says Peter. “Seb embodies what cross-disciplinary science at the Broad is about. He brought his mathematical understanding, sat with biologists for a while, didn’t become a biologist — but learned enough to tackle the problem differently.”
Peter recalls that the hierarchical insight emerged after Seb had spent time absorbing how biologists talk about cell types. “He realized life isn’t organized in a flat linear context — it’s hierarchical. And if you teach a model that structure, it performs better.”
For Lorin, Seb’s growth mindset has been equally important. “He’s always willing to put himself in a place where he might feel like a fish out of water,” he says. “That humility and eagerness to learn allows him to grow incredibly fast.”
Seb emphasizes the mentorship environment. “They were open to me working on what I found interesting and were very supportive — even when ideas didn’t work. In science, you have many ideas that fail before one works. Seeing how mentors react when things don’t work tells you a lot.”
Lorin and Peter both agree that Seb’s collaboration with Project Ex Vivo is a great model for future postdoctoral fellows at the Schmidt Center who want to work with them, with the freedom to try new ideas, utilizing the diversity of skill sets that the group has.
Seb presenting at the Schmidt Center Symposium 2025
What are you working on now?
Seb is now excited to think about evaluation — how do we rigorously compare AI systems in biology?
“In images or text, you can often tell when something looks wrong,” he says. “But if a model predicts the outcome of a biological experiment, it’s much harder to have that immediate intuition.”
He’s interested in defining meaningful mathematical metrics to compare models, especially as new ones are released rapidly.
“In the end, we’re comparing sets of numbers,” he says. “The question is: what’s a rigorous and interesting way to do that?”
Long term, he’s also thinking about efficiency.
“I’m interested in making these AI models more efficient, from a cost or energy perspective,” he says. “That matters in biology, but also more broadly as AI becomes part of everyday life.”
What has your experience at the Schmidt Center — and at the Broad more broadly — been like?
“Incredibly rewarding,” Seb says. “Having fellows with varied backgrounds in applied mathematics, computer science, ML, and computational biology on the same floor makes the coffee chats much more interesting.”
He especially values the weekly group meetings at the Schmidt Center.
“You get challenged by people who think about computational methods from different angles,” he says. “If you stay in one group and you’re the one computational expert on a topic, it’s harder to get that kind of feedback. Here, you have people approaching related problems from different perspectives, and that’s incredibly productive.”
Seb presenting an MIA flash talk
Beyond the Schmidt Center, Seb is also part of the Models, Inference & Algorithms (MIA) steering committee — a seminar series that brings together biologists and machine learning researchers, which he was following even before he joined the Broad.
“MIA was something I really enjoyed as a listener,” he says. “I particularly liked the primers, where someone takes the time to slowly dig into the important details of an algorithm. Coming from a different field, that was incredibly valuable for me.”
After joining the Broad, he became involved in organizing seminars and inviting speakers.
“It’s been a very fun and energizing experience,” he says. “You learn from the speakers, from their students and postdocs — and it also feels like a way to give back to the community.” (Learn more about Seb’s role and thoughts on MIA in our retrospective video – MIA: 10 Years of Models, Inference & Algorithms (MIA) at Broad Institute.)
Together, these spaces — the Schmidt Center, Project Ex Vivo, and MIA — have given Seb what he values most: a diverse intellectual environment where mathematical thinking and biological questions meet.
The Schmidt Center is thrilled to have such an intelligent and creative postdoc!
What advice would you give aspiring researchers?
“If you hear about a project that genuinely interests you, go after it,” Seb says. “Research can feel daunting because you’re left with your own ideas. But that’s also what makes it exciting.”
He encourages students to try research early and trust their curiosity.
“Trends come and go,” he says. “It’s better to follow what you actually find fun.”
What do you enjoy outside of research?
Sports have always been central to Seb’s life. He grew up playing basketball and now enjoys racket sports like tennis and paddle. As a first-time resident of the U.S., he’s also been exploring New England and hopes to see more of the United States.
Chitra received his PhD in computer science from Princeton University. Before joining Hopkins, he was a postdoctoral fellow at the Eric and Wendy Schmidt Center at the Broad Institute of MIT and Harvard and a software engineer at Facebook.
Tell us a little bit about your research.
I work in computational biology—specifically, developing algorithms for analyzing and interpreting large-scale biological data. Numerous technological breakthroughs over the past two decades—ranging from high-throughput DNA sequencing to single-cell/spatial genomics and CRISPR gene editing—have enabled scientists to measure diverse molecular modalities (DNA, RNA, proteins, etc.) across many biological systems (e.g., the brain or a tumor). However, existing machine learning (ML) frameworks often cannot be directly applied in this area because of the technical noise, sparsity, heterogeneity, and other unique aspects of biological data. As such, my research draws on tools from statistics, geometry, and graph theory to develop specialized algorithms for high-dimensional and multimodal biological data, with the broad goal of understanding biology at the molecular and cellular level.
A topographic map of the mouse cerebellum learned by deep learning algorithm GASTON. Each dot is a single cell; the color of a cell indicates the “isodepth,” a quantity analogous to the elevation in a topographic map of a landscape. Cells with equal isodepth (shown as contour lines) have similar gene expression profiles.Type image caption here (optional).
Tell us about a project you are excited about.
I’m particularly excited by new computational problems in spatial biology. Recent “spatial transcriptomics” technologies measure both the gene expression and spatial location of individual cells, enabling us to understand how cells are organized and interact with one another inside our tissues. These datasets are also extremely sparse and high-dimensional, which creates unique mathematical and computational challenges. How do you model both the spatial geometry of cells and the high-dimensional geometry of gene expression measurements? How do you identify meaningful spatial patterns (e.g., continuous gradients) when there is such large data sparsity?
To address these challenges, we’ve developed new deep learning methods for learning “topographic maps” of tissues. Our topographic maps are analogous to elevation maps in geography, but instead of elevation, they describe a quantity called “isodepth” that reveals both the spatial organization of cell types and the spatial gradients of gene expression across a tissue, allowing us to visually “see” how tissues are organized. Mathematically, our topographic maps are based on a new model of spatial gradients in sparse data.
Why this? What drives your passion for your field?
While I’m now in a computer science department, I’ve always loved the process of doing math: experimenting, devising, and mulling over my conjectures before writing up my arguments in a rigorous, airtight proof. But after two years of taking pure math classes in college, I realized that I ultimately wanted to do work with more real-world applications. By pure coincidence, I did a summer research internship with a former mathematician who moved into computational biology; I enjoyed the experience so much that I stayed in the field and did my PhD with him. I like working in computational biology because I get to develop mathematical frameworks and ML algorithms that address fundamental problems in biology, thus marrying my love of math with my desire to create a tangible impact with my work.
One especially exciting part of working in computational biology is how fast the field evolves. New biological technologies are constantly being developed, and each new technology brings new kinds of data and new biological questions that existing methods can’t quite answer. It’s fun being in a field where the problems aren’t fully defined yet and where developing novel mathematical ideas and ML algorithms directly impacts how we understand biology.
What classes are you teaching?
This semester, I’m teaching a graduate course on advanced topics in single-cell and spatial biology. In this class, students read and discuss research papers on computational methods and ML algorithms for single-cell and spatial genomics data, covering topics such as clustering, dimensionality reduction, optimal transport, and foundation models. In addition to teaching students how to read research papers, I also hope to teach them how to translate biological problems into clear and rigorously defined computational problems.
Why are you excited to be joining the Johns Hopkins Department of Computer Science?
Johns Hopkins is an incredibly exciting place to work at the intersection of biology and AI/ML. The university is a powerhouse in biomedical research—its Schools of Medicine and Public Health are among the best in the world—and researchers here are constantly developing exciting new experimental technologies and generating rich biological datasets. Even in the few months that I’ve been here, conversations with experimental biologists have been a major source of motivation for my own research. At the same time, Hopkins has a growing and vibrant AI/ML community, with the department hiring many strong faculty through the Data Science and AI Institute. I’m very lucky that all of these exceptional researchers are my colleagues!
I’m also excited to be working in a department with such strong students. Hopkins students at all stages—undergraduate, master’s, and PhD—are exceptionally strong, curious, and motivated; I’m looking forward to mentoring and collaborating with them.
Besides your work, what are some of your other hobbies and passions?
I enjoy rock climbing, specifically bouldering. Since moving to Baltimore, I’ve been climbing regularly at a nearby gym and exploring local outdoor bouldering spots in Maryland. The climbing community here is very welcoming!
This article was originally written by and for the Johns Hopkins Whiting School of Engineering.
While technologies like Perturb-seq let scientists observe cells' responses to genetic changes at scale, the ability to predict those responses using machine learning tools could greatly accelerate research on disease mechanisms.
Former Schmidt Center MEng Emily Liu, current PhD fellow Jiaqi Zhang, and Director Caroline Uhler have developed a new model called Single Cell Causal Variational Autoencoder (SCCVAE) that addresses two key challenges in modeling unseen genetic perturbations: lack of generalizability, and difficulty with noisy, large-scale single cell data. By integrating deep learning and mechanistic approaches, SCCVAE allows the team to simulate new experiments and reveal groups of genes that work in concert.
Many data types are used to diagnose and treat disease, but interpreting them can be challenging. Scientists in the Eric and Wendy Schmidt Center and Data Sciences Platform built an AI framework, MODES (Multi-mOdal Disentangled Embedding Space), that decouples or separates the information that is shared in all modalities from those that only one test can uniquely measure. They applied MODES to a cardiovascular model using ECG and cardiac MRI and showed that it provides a better picture of an individual’s health than previous models, which may help improve diagnostics.
The authors of the paper are Schmidt Center postdoctoral fellow Sana Tonekaboni, Senior Group Leader and Prinicipal Machine Learning Scientist Sam Freesun Friedman, former Schmidt Center PhD fellow Xinyi Zhang, Director of Machine Learning for Health Mahnaz Maddah, and Schmidt Center Director Caroline Uhler.
What is patient privacy for? The Hippocratic Oath, thought to be one of the earliest and most widely known medical ethics texts in the world, reads: “Whatever I see or hear in the lives of my patients, whether in connection with my professional practice or not, which ought not to be spoken of outside, I will keep secret, as considering all such things to be private.”
As privacy becomes increasingly scarce in the age of data-hungry algorithms and cyberattacks, medicine is one of the few remaining domains where confidentiality remains central to practice, enabling patients to trust their physicians with sensitive information.
But a paper co-authored by MIT researchers investigates how artificial intelligence models trained on de-identified electronic health records (EHRs) can memorize patient-specific information. The work, which was recently presented at the 2025 Conference on Neural Information Processing Systems (NeurIPS), recommends a rigorous testing setup to ensure targeted prompts cannot reveal information, emphasizing that leakage must be evaluated in a health care context to determine whether it meaningfully compromises patient privacy.
Foundation models trained on EHRs should normally generalize knowledge to make better predictions, drawing upon many patient records. But in “memorization,” the model draws upon a singular patient record to deliver its output, potentially violating patient privacy. Notably, foundation models are already known to be prone to data leakage.
“Knowledge in these high-capacity models can be a resource for many communities, but adversarial attackers can prompt a model to extract information on training data,” says Sana Tonekaboni, a postdoc at the Eric and Wendy Schmidt Center at the Broad Institute of MIT and Harvard and first author of the paper. Given the risk that foundation models could also memorize private data, she notes, “this work is a step towards ensuring there are practical evaluation steps our community can take before releasing models.”
To conduct research on the potential risk EHR foundation models could pose in medicine, Tonekaboni approached MIT Associate Professor Marzyeh Ghassemi, who is a principal investigator at the Abdul Latif Jameel Clinic for Machine Learning in Health (Jameel Clinic), a member of the Computer Science and Artificial Intelligence Lab. Ghassemi, a faculty member in the MIT Department of Electrical Engineering and Computer Science and Institute for Medical Engineering and Science, runs the Healthy ML group, which focuses on robust machine learning in health.
Just how much information does a bad actor need to expose sensitive data, and what are the risks associated with the leaked information? To assess this, the research team developed a series of tests that they hope will lay the groundwork for future privacy evaluations. These tests are designed to measure various types of uncertainty, and assess their practical risk to patients by measuring various tiers of attack possibility.
“We really tried to emphasize practicality here; if an attacker has to know the date and value of a dozen laboratory tests from your record in order to extract information, there is very little risk of harm. If I already have access to that level of protected source data, why would I need to attack a large foundation model for more?” says Ghassemi.
With the inevitable digitization of medical records, data breaches have become more commonplace. In the past 24 months, the U.S. Department of Health and Human Services has recorded 747 data breaches of health information affecting more than 500 individuals, with the majority categorized as hacking/IT incidents.
Patients with unique conditions are especially vulnerable, given how easy it is to pick them out. “Even with de-identified data, it depends on what sort of information you leak about the individual,” Tonekaboni says. “Once you identify them, you know a lot more.”
In their structured tests, the researchers found that the more information the attacker has about a particular patient, the more likely the model is to leak information. They demonstrated how to distinguish model generalization cases from patient-level memorization, to properly assess privacy risk.
The paper also emphasized that some leaks are more harmful than others. For instance, a model revealing a patient’s age or demographics could be characterized as a more benign leakage than the model revealing more sensitive information, like an HIV diagnosis or alcohol abuse.
The researchers note that patients with unique conditions are especially vulnerable given how easy it is to pick them out, which may require higher levels of protection. “Even with de-identified data, it really depends on what sort of information you leak about the individual,” Tonekaboni says. The researchers plan to expand the work to become more interdisciplinary, adding clinicians and privacy experts as well as legal experts.
“There’s a reason our health data is private,” Tonekaboni says. “There’s no reason for others to know about it.”
This work supported by the Eric and Wendy Schmidt Center at the Broad Institute of MIT and Harvard, Wallenberg AI, the Knut and Alice Wallenberg Foundation, the U.S. National Science Foundation (NSF), a Gordon and Betty Moore Foundation award, a Google Research Scholar award, and the AI2050 Program at Schmidt Sciences. Resources used in preparing this research were provided, in part, by the Province of Ontario, the Government of Canada through CIFAR, and companies sponsoring the Vector Institute.
This article was originally written for the MIT Schwarzman College of Computing.
Winning the opening phase of the Autoimmune Disease Machine Learning Challenge marked a major milestone for ETH Zürich PhD student Kalin Nonchev, and a strong start to an ambitious global competition. “This challenge gave me the opportunity to test our methods in a completely different setting – autoimmune disease instead of cancer,” he said. “It was very rewarding but challenging, and it highlighted the importance of building flexible, modular models that can be efficiently adapted to new disease contexts.” His model would go on to place second in the next phase, Crunch 2, further fortifying his role in a competition that drew nearly 1,000 participants from 62 countries. To learn more about the top participants’ experiences – keep reading below.
Launched on October 28, 2024 by the Eric and Wendy Schmidt Center and the Klarman Cell Observatory (KCO) at the Broad Institute and hosted on the CrunchDAO platform, the three-part challenge aimed to improve the diagnosis of inflammatory bowel disease (IBD) by applying machine learning to real biological data. Participants built models that integrated spatial transcriptomics with histopathology images, tools that could eventually enable earlier detection and personalized treatment for patients with chronic conditions.
Watch an overview of the Autoimmune Disease ML Challenge
“It’s exciting to see these challenges become an annual tradition that brings together interdisciplinary teams from around the world,” said Caroline Uhler, director of the Schmidt Center and the Andrew (1956) and Erna Viterbi Professor of Engineering at MIT. “What sets our challenges apart is not just the scale or the scientific ambition, but the fact that we follow through with experimental validation. The challenges are powerful because they bridge computational predictions and biological testing.”
“This challenge demonstrates how introducing the global machine learning community to a biological problem can accelerate scientific and clinical discoveries,” added Ramnik Xavier, director of the KCO, gastroenterologist at Massachusetts General Hospital, and Kurt J. Isselbacher Professor of Medicine at Harvard Medical School. “Looking beyond the boundaries of one domain reveals opportunities we wouldn’t find alone.”
The challenge consists of three parts, or Crunches. In Crunch 1, participants predicted gene expression in spatial transcriptomics data from matched pathology images and in Crunch 2, they predicted unseen genes. Finally, in Crunch 3, participants identified gene markers for pre-cancerous regions. The gene panels developed by top performers will be tested in patient samples through lab experiments at the Broad (see the full, detailed specifications here).
Photo Credits: Jean Herelle
CrunchDAO, a data science competition platform, hosted the challenge. “The beauty of ML challenges is that they bring a community together – collective intelligence has immense potential and can lead to progress much faster than an individual working alone,” said Jean Herelle, founder and CEO of CrunchDAO. “CrunchDAO was really excited to partner with the Schmidt Center to bring a new type of data science problem with a biological focus to our community. It was a perfect fit because, like our other challenges, this one had a purpose-driven mission.”
Meet the Top Performers
Top-performing teams approached the task from a range of backgrounds and perspectives, but they shared a few key strategies.
Kalin Nonchev (1st in Crunch 1, 2nd in Crunch 2)
For Kalin Nonchev, the Autoimmune Disease Machine Learning Challenge wasn’t just about building a high-performing model – it was also an opportunity to push his research into new terrain. With a background in computer science and bioinformatics, Kalin had been developing DeepSpot, a multimodal framework for spatial transcriptomics, in the context of cancer.
“Adapting DeepSpot to this new environment involved hands-on troubleshooting and provided deeper insight into the unique characteristics of spatial transcriptomics across diverse tissue regions, particularly in complex diseases like inflammatory bowel disease,” he said. “The histology images, spatial structure, and technical artifacts differed significantly from what we had trained on before.”
What stood out most to Kalin, though, was the community. “The challenge drew competitors from around the world, fostering a friendly atmosphere despite the rivalry, and it was genuinely fun,” Kalin said. “It was clear that everyone shared a strong passion for advancing spatial biology methods. Having an independent platform like CrunchDAO to test ideas, compare methods, and see where your approach stands was extremely motivating.”
Manfred Seiwald (2nd in Crunch 1)
Manfred Seiwald, a senior software developer in the Department of Biosciences and Medical Biology at the University of Salzburg, secured second place in Crunch 1. Similarly to most top performing models in Crunch 1, his model leveraged the embedding of a histopathology foundation model as an image encoder. A key feature of his approach was the use of shared decoders trained jointly on histopathology images and gene expression data to predict gene expression. This approach allowed his model to effectively align visual and molecular features without the need for complex adaptations.
Team PathBio (3rd in Crunch 1)
Team PathBio brought together a group of deep learning researchers: Sen Yang (team lead, Medical and Health Laboratory, Ant Group), Jinxi Xiang (Stanford University), Wei Yuan (Sichuan University), Yijiang Chen (Stanford University), and Xiyue Wang (Stanford University).
Though experienced in medical image analysis, none of the team members had worked with spatial biology data before. “This challenge offered us invaluable insights into spatial biology, significantly expanding our understanding beyond our prior expertise in deep learning and medical image analysis,” they said. “One particularly impactful aspect was discovering the intricate relationship between tissue morphology and spatial gene expression patterns.”
That realization shaped their modeling strategy, which captured spatial context by applying a successive attention mechanism and gaussian masking to small patches of the histopathology images. “We observed that spatially adjacent tissue pixels often harbor similar cell populations, thereby exhibiting similar gene expression profiles,” they explained.
The experience inspired the team’s broader goals. “We picked up a wealth of knowledge in bioinformatics during the hands-on experimentations of the challenge, from processing complex genomic datasets to understanding the biological context behind autoimmune disease markers,” they said. “The nature of spatial biology data co-registered with digital pathology data at the pixel level has opened a lot of doors and sparked many new ideas for us as future research directions, and we can’t wait to experiment and explore these ideas.”
Alexis Gassmann – Tarandros (4th in Crunch 1, 1st in Crunch 2)
Alexis Gassmann, a freelance data scientist and organic farmer, stood out in both phases of the challenge with a sophisticated modeling pipeline that combined modern machine learning techniques. He was the only participant who applied contrastive learning to learn a shared latent space between image and molecular data. He also employed a mixture-of-experts approach – a flexible and scalable machine learning technique that relies on multiple "expert" subnetworks trained to handle different parts of a problem – and a spatial coordinate attention mechanism, enabling the model to capture both local cell organization and global image features.
Alexis ranked among the top performers in Crunch 1. In Crunch 2, his contrastive learning, similarity-based method allowed for accurate cross-modal gene expression transfer from single-cell to spatial context, ultimately earning him first place.
Expanding on his earlier work was a key component for helping Alexis reach the top. “It was demanding but highly rewarding,” he said. “The challenge was structured in three parts that built on each other, making it feel like solving a solution step by step. I enjoyed designing a solution that progressed from predicting specific gene expression from histology images then generalizing across the full transcriptome and using them to identify early biomarkers of colorectal cancer.”
His success across both phases demonstrated the power of combining deep representation learning with biologically informed structure – an approach that proved both adaptable and effective across distinct modeling tasks.
Marios Gavrielatos and Konstantinos Kyriakidis (5th in Crunch 1, 3rd in Crunch 2)
Marios Gavrielatos, a researcher at the Mayo Clinic, and Konstantinos Kyriakidis, a researcher at UC Santa Cruz, returned to this competition after winning the Cancer Immunotherapy Machine Learning Competition in 2023. This time, they brought a hybrid model that combined convolutional (CNN) and fully connected networks (MLP) to analyze spatial gene expression in IBD.
In Crunch 1, their model enhanced embeddings from pretrained histopathology models to make gene-level predictions by incorporating the statistical properties of the outputs to refine accuracy. For Crunch 2, they developed a multi-channel CNN that encoded structured input from five nearest neighbors in single-cell space, followed by an MLP to generalize to genes unseen in training. This architecture captured local spatial relationships, while preserving flexibility across gene sets.
For both Marios and Konstantinos, the challenge offered a chance to sharpen their research skills and apply their training in new ways. “This year's challenge allowed me to fully demonstrate my research capabilities,” said Marios. “I had significantly more time to dedicate compared to last year... working from the ground up. Successfully contributing to our top-performing solution validated my ability to rapidly master new domains.”
Konstantinos added: “The point of these competitions, at least for me, is to expand the current boundaries of thinking... Every challenge adds a new layer of knowledge to my thinking toolbox.”
They also credited their biology backgrounds – Marios with degrees in biology and biomedical data science and Konstantinos with degrees in pharmaceutical sciences – for helping them bridge the machine learning and biological domains.
“These challenges showcase, once again, the importance of interdisciplinary collaboration,” they said. “People with different backgrounds and experiences work together and create a unique flow of ideas that sometimes can be transformative. We hope we see more challenges like these in the future!”
Scientific Takeaways from the First Two Phases
Despite the complexity of the task, common trends emerged from the top-performing submissions. The submissions are being analyzed by Schmidt Center scientists, including postdoctoral fellows Kai Cao and Luezhen Yuan, computational scientist Rajshikhar Gupta, and director of computational biology at the KCO Orr Ashenberg.
High-performing models in Crunch 1 commonly utilize foundational models trained on vast histopathology image datasets like UNI, TIAToolbox, Path etc. to derive meaningful representations of the images. They then align single-cell gene expression and histopathology imaging data into a shared representation. Some successful methods also incorporate the spatial arrangement of cells through positional encoding or self-attention techniques.
"While the difference in performance between ranked models is modest, all of them significantly outperform the baseline,” said Raj.
The performance of Crunch 2 depends heavily on the accuracy of Crunch 1, as it takes Crunch 1’s predictions as input. Most existing methods for Crunch 2 follow relatively simple strategies, which can be broadly classified into two main categories: similarity-based methods and MLP-based methods.
The similarity-based methods compute the similarity between spatial and single-cell data either directly, or after alignment of the two modalities in a shared latent space, based on the genes provided in Crunch 1. After establishing neighbor relationships, they predict the expression of genes unseen in Crunch 1 by transferring expression from single-cell to spatial data using weighted averages. In contrast, MLP-based methods train a neural network on single-cell data to map from the genes seen in training to the unseen genes. Once trained, the model takes Crunch 1’s predictions as input and outputs the imputed expression for the unseen genes.
"It's remarkable how, despite the simplicity of the design, the participating models proved to be highly effective, particularly in predicting highly expressed genes,” said Kai.
“These top models seem to have potential in predicting a cell type's spatial distribution and other more demanding downstream tasks from H&E images alone," added Luezhen.
The use of these diverse but complementary strategies shows how a variety of machine learning approaches can converge toward solving real biological challenges.
What’s Next
While Kai and Raj continue the formal benchmarking of top methods in Crunch 1 and Crunch 2, other team members, including Luezhen and Orr, are analyzing the Crunch 3 submissions.
“We are in the process of preparing gene panels based on the participants’ proposed potential markers of dysplasia for experimental validation at the Broad Institute,” said Orr. “Experimental validation is an important part of the challenge and represents a key step toward clinical translation, turning model predictions into potential diagnostic tools for IBD and related diseases.”
Stay tuned for updates on the final results, as well as the launch of our next machine learning challenge – the second in our Cell Perturbation Prediction Challenge series, which will focus on shifting adipocyte cell states in vitro.
Analyzing how variables within massive datasets rise and fall in relation to each other can reveal hidden structures in processes like gene expression. The methods for studying such dependencies, however, often focus on linear relationships or don't scale to millions of samples and tens of thousands of variables.
In response, Adit Radhakrishnan, Yajit Jain, Caroline Uhler, and Eric Lander have developed the InterDependence Score (IDS), a computationally light algorithm that uncovers relationships in large datasets that elude other correlation measures, such as complex expression patterns underlying cellular programs and states. It also, they found, provides fundamental insights into neural networks' predictive capabilities.
Radhakrishnan is a former Eric and Wendy Schmidt Center postdoctoral fellow and current assistant professor at MIT; Jain is a former Schmidt Center postdoctoral fellow and current Senior ML Scientist at the Lander Lab; Uhler is the director of the Schmidt Center and the Andrew and Erna Viterbi Professor of Engineering at MIT; and Lander is the founding director and a core institute member of the Broad Institute.
Although outbreaks of Ebola virus are rare, the disease is severe and often fatal, with few treatment options. Rather than targeting the virus itself, one promising therapeutic approach would be to interrupt proteins in the human host cell that the virus relies upon. However, finding those regulators of viral infection using existing methods has been difficult and is especially challenging for the most dangerous viruses like Ebola that require stringent high-containment biosafety protocols.
Now, researchers at the Broad Institute and the National Emerging Infectious Diseases Laboratories (NEIDL) at Boston University have used an image-based screening method developed at the Broad to identify human genes that, when silenced, impair the Ebola virus’s ability to infect. The method, known as optical pooled screening (OPS), enabled the scientists to test, in about 40 million CRISPR-perturbed human cells, how silencing each gene in the human genome affects virus replication.
Using machine-learning-based analyses of images of perturbed cells, they identified multiple host proteins involved in various stages of Ebola infection that when suppressed crippled the ability of the virus to replicate. Those viral regulators could represent avenues to one day intervene therapeutically and reduce the severity of disease in people already infected with the virus. The approach could be used to explore the role of various proteins during infection with other pathogens, as a way to find new drugs for hard-to-treat infections.
“This study demonstrates the power of OPS to probe the dependency of dangerous viruses like Ebola on host factors at all stages of the viral life cycle and explore new routes to improve human health,” said co-senior author Paul Blainey, a Broad core faculty member and professor in the Department of Biological Engineering at MIT.
Previously, members of the Blainey lab developed the optical pooled screening method as a way to combine the benefits of high-content imaging, which can show a range of detailed changes in large numbers of cells at once, with those of pooled perturbational screens, which show how genetic elements influence these changes. In this study, they partnered with the laboratory of Robert Davey at BU to apply optical pooled screening to Ebola virus.
The team used CRISPR to knock out each gene in the human genome, one at a time, in nearly 40 million human cells, and then infected each cell with Ebola virus. They next fixed those cells in place in laboratory dishes and inactivated them, so that the remaining processing could occur outside of the high-containment lab.
After taking images of the cells, they measured overall viral protein and RNA in each cell using the CellProfiler image analysis software, and to get even more information from the images, they turned to AI. With help from team members in the Eric and Wendy Schmidt Center at the Broad, led by study co-author and Broad core faculty member and Schmidt Center director Caroline Uhler, they used a deep learning model to automatically determine the stage of Ebola infection for each single cell. The model was able to make subtle distinctions between stages of infection in a high-throughput way that wasn’t possible using prior methods.
“The work represents the deepest dive yet into how Ebola virus rewires the cell to cause disease, and the first real glimpse into the timing of that reprogramming,” said co-senior author Robert Davey, director of the National Emerging Infectious Diseases Laboratories at Boston University, and professor of microbiology at BU Chobanian and Avedisian School of Medicine. “AI gave us an unprecedented ability to do this at scale.”
By sequencing parts of the CRISPR guide RNA in all 40 million cells individually, the researchers determined which human gene had been silenced in each cell, indicating which host proteins (and potential viral regulators) were targeted. The analysis revealed hundreds of host proteins that, when silenced, altered overall infection level, including many required for viral entry into the cell.
Knocking out other genes enhanced the amount of virus within inclusion bodies, structures that form in the human cell to act as viral factories, and prevented the infection from progressing further. Some of these human genes, such as UQCRB, pointed to a previously unrecognized role for mitochondria in the Ebola virus infection process that could possibly be exploited therapeutically. Indeed, treating cells with a small molecule inhibitor of UQCRB reduced Ebola infection with no impact on the cell’s own health.
Other genes, when silenced, altered the balance between viral RNA and protein. For example, perturbing a gene called STRAP resulted in increased viral RNA relative to protein. The researchers are currently doing further studies in the lab to better understand the role of STRAP and other proteins in Ebola infection and whether they could be targeted therapeutically.
In a series of secondary screens, the scientists examined some of the highlighted genes’ roles in infection with related filoviruses. Silencing some of these genes interrupted replication of Sudan and Marburg viruses, which have high fatality rates and no approved treatments, so it’s possible a single treatment could be effective against multiple related viruses.
The study’s approach could also be used to examine other pathogens and emerging infectious diseases and look for new ways to treat them.
“With our method, we can measure many features at once and uncover new clues about the interplay between virus and host, in a way that’s not possible through other screening approaches,” said co-first author Rebecca Carlson, a former graduate researcher in the labs of Blainey and Nir Hacohen at the Broad and who co-led the work along with co-first author J.J. Patten at Boston University.
This work was funded in part by the Broad Institute, the National Human Genome Research Institute, the Burroughs Wellcome Fund, the Fannie and John Hertz Foundation, the National Science Foundation, the George F. Carrier Postdoctoral Fellowship, the Eric and Wendy Schmidt Center at the Broad Institute, the National Institutes of Health, and the Office of Naval Research.
The following article was issued today by the Broad Institute of MIT and Harvard.
Before graduating with her bachelor’s degree in Artificial Intelligence and Decision Making from MIT, Yoanna Turura completed an ambitious step that few students experience so early in their careers: presenting original research at CHIL 2025, a leading conference at the intersection of machine learning and healthcare. Her project, called Latentverse, is a new open-source benchmarking toolkit for evaluating self-supervised learned representations in clinical data. It began during her time as a fellow at the Eric and Wendy Schmidt Center, shaped by the mentorship of Schmidt Center postdoctoral fellow Sana Tonekaboni.
Their paper, The Latentverse: An Open-Source Benchmarking Toolkit for Evaluating Latent Representations (Yoanna Turura, Sam Freesun Friedman, Aurora Cremer, Mahnaz Maddah, and Sana Tonekaboni), was accepted to CHIL 2025 – the 6th Annual Conference on Health, Inference, and Learning by the Association for Health Learning and Inference, held in June 2025 at UC Berkeley. For Yoanna, who had never submitted a paper before, the experience was a defining milestone and a chance to bring fresh ideas to a global community of researchers.
“Yoanna is a responsible and creative researcher,” said Sana. “She’s great at coming up with new ideas and then actually making them happen. While she's open to guidance, she often takes the lead, exploring new paths and developing her own unique methods, which really highlights her independent thinking. I really enjoyed working with her!”
We spoke with Yoanna before and after the conference to learn more about her work, how she prepared for her first major presentation, and how this experience will shape her next steps as a researcher.
---------
First, we caught up with Yoanna as she planned to attend CHIL.
Congratulations on your CHIL paper! How does it feel to have your work selected, especially as an undergraduate student?
I was really excited! This was my first time submitting a paper to a conference, and we had been working on the project since the beginning of the school year. It felt great to see our work recognized as useful to the field. As an early-career researcher, I’m thrilled I could contribute to something that could help others – and I can’t wait to share it with the community.
What are you most excited about at CHIL this year?
It’s my first time attending CHIL, and I’m eager to learn more about new, fascinating developments at the intersection of health and machine learning – an area I’m passionate about. I’m also looking forward to the roundtables, which I think will spark great dialogue and idea-sharing.
Tell us more about your work.
Self-supervised models can create powerful embeddings, but evaluating their quality remains a challenge as most tools are domain-specific, lack standardized metrics, or require raw data. We created Latentverse, an open-source library and web app that evaluates latent representations using a suite of interpretable tests, including probing, clustering, disentanglement, expressiveness, and robustness. It’s designed to help researchers compare and improve representation learning methods across domains.
How has Sana guided you?
Sana has been an incredible mentor during my first research experience. She was instrumental in my development and supported me from day one. Besides meeting regularly, she made herself available during any blockers that I had, helped with literature reviews, and provided detailed feedback as I created the framework. She made my experience more meaningful by bringing me to the broader Machine Learning for Health (ML4H) team, where I could brainstorm and present my ideas.
Sana encouraged me to submit my first paper on our joint work, and she was there through every step of the process – even late nights working together. I’ve learned so much from her skillful guidance, and I’m deeply inspired by her dedication to both the field and her mentees.
---------
Then, we chatted with Yoanna once she returned from the conference.
What were some of your highlights from CHIL?
I loved the panel discussions and Q&As. Coming from vastly different backgrounds, the speakers brought unique perspectives from across healthcare and focused on practical applications, like responsible AI integration or better evaluation for GenAI models. It was inspiring to hear candid reflections on what’s working, what’s not, and where the field is headed. I also learned about some groundbreaking, innovative tools that are being developed to improve the clinician and patient experience.
How did the poster presentation go?
It went really well! Lots of people stopped by to learn about Latentverse and they were curious about how it could be applied to their projects. Many had thoughtful questions about how to interpret the scores and visualizations, and some of that feedback has already sparked ideas for improving the web app. It was exciting to see people scan the QR code to the Latentverse library and want to use it in their own work. I was really glad to explain this tool to others.
Any recommendations for future undergraduates presenting at CHIL?
CHIL is all about bringing together people from diverse healthcare backgrounds (clinicians, entrepreneurs, computer scientists, and so on) to exchange ideas, share solutions, ideate how to advance the field, and collaborate. So don’t limit yourself to poster sessions – take advantage of the roundtables, coffee breaks, and lunch sessions to talk about your work and learn from others. CHIL encourages this kind of sharing and there is no better place for thoughtful conversation and community-building when everyone is united for a common purpose.
Yoanna will be starting as a software engineer at Amazon this summer.
MIT researchers have developed a new theoretical framework for studying the mechanisms of treatment interactions. Their approach allows scientists to efficiently estimate how combinations of treatments will affect a group of units, such as cells, enabling a researcher to perform fewer costly experiments while gathering more accurate data.
As an example, to study how interconnected genes affect cancer cell growth, a biologist might need to use a combination of treatments to target multiple genes at once. But because there could be billions of potential combinations for each round of the experiment, choosing a subset of combinations to test might bias the data their experiment generates.
In contrast, the new framework considers the scenario where the user can efficiently design an unbiased experiment by assigning all treatments in parallel, and can control the outcome by adjusting the rate of each treatment.
The MIT researchers theoretically proved a near-optimal strategy in this framework and performed a series of simulations to test it in a multiround experiment. Their method minimized the error rate in each instance.
This technique could someday help scientists better understand disease mechanisms and develop new medicines to treat cancer or genetic disorders.
Co-lead author, Schmidt Center PhD fellow, and MIT EECS graduate student Jiaqi Zhang
“We’ve introduced a concept people can think more about as they study the optimal way to select combinatorial treatments at each round of an experiment. Our hope is this can someday be used to solve biologically relevant questions,” says graduate student Jiaqi Zhang, an Eric and Wendy Schmidt Center Fellow and co-lead author of a paper on this experimental design framework.
She is joined on the paper by co-lead author Divya Shyamal, an MIT master's engineering student; and senior author Caroline Uhler, the Andrew and Erna Viterbi Professor of Engineering in EECS and the MIT Institute for Data, Systems, and Society (IDSS), who is also director of the Eric and Wendy Schmidt Center and a researcher at MIT’s Laboratory for Information and Decision Systems (LIDS). The research was recently presented at the International Conference on Machine Learning.
Simultaneous treatments
Treatments can interact with each other in complex ways. For instance, a scientist trying to determine whether a certain gene contributes to a particular disease symptom may have to target several genes simultaneously to study the effects.
To do this, scientists use what are known as combinatorial perturbations, where they apply multiple treatments at once to the same group of cells.
“Combinatorial perturbations will give you a high-level network of how different genes interact, which provides an understanding of how a cell functions,” Zhang explains.
Since genetic experiments are costly and time-consuming, the scientist aims to select the best subset of treatment combinations to test, which is a steep challenge due to the huge number of possibilities.
Picking a suboptimal subset can generate biased results by focusing only on combinations the user selected in advance.
The MIT researchers approached this problem differently by looking at a probabilistic framework. Instead of focusing on a selected subset, each unit randomly takes up combinations of treatments based on user-specified dosage levels for each treatment.
The user sets dosage levels based on the goal of their experiment — perhaps this scientist wants to study the effects of four different drugs on cell growth. The probabilistic approach generates less biased data because it does not restrict the experiment to a predetermined subset of treatments.
The dosage levels are like probabilities, and each cell receives a random combination of treatments. If the user sets a high dosage, it is more likely most of the cells will take up that treatment. A smaller subset of cells will take up that treatment if the dosage is low.
“From there, the question is how do we design the dosages so that we can estimate the outcomes as accurately as possible? This is where our theory comes in,” Shyamal adds.
Co-lead author, Schmidt Center fellow, and MIT master's of engineering student Divya Shyamal
Their theoretical framework shows the best way to design these dosages so one can learn the most about the characteristic or trait they are studying.
After each round of the experiment, the user collects the results and feeds those back into the experimental framework. It will output the ideal dosage strategy for the next round, and so on, actively adapting the strategy over multiple rounds.
Optimizing dosages, minimizing error
The researchers proved their theoretical approach generates optimal dosages, even when the dosage levels are affected by a limited supply of treatments or when noise in the experimental outcomes varies at each round.
In simulations, this new approach had the lowest error rate when comparing estimated and actual outcomes of multiround experiments, outperforming two baseline methods.
In the future, the researchers want to enhance their experimental framework to consider interference between units and the fact that certain treatments can lead to selection bias. They would also like to apply this technique in a real experimental setting.
“This is a new approach to a very interesting problem that is hard to solve. Now, with this new framework in hand, we can think more about the best way to design experiments for many different applications,” Zhang says.
This research is funded, in part, by the Advanced Undergraduate Research Opportunities Program at MIT, Apple, the National Institutes of Health, the Office of Naval Research, the Department of Energy, the Eric and Wendy Schmidt Center at the Broad Institute, and a Simons Investigator Award.
This article was originally written for and posted on MIT News.
When the first sequence of the human genome project was drafted 25 years ago, few could have predicted how transformative it would be for biomedicine. "Today's young researchers cannot imagine doing biomedical research without having the foundation of the human genome," said Todd Golub, Director and Founding Core Institute Member of the Broad Institute of MIT and Harvard, in his opening remarks. Golub compared that turning point from the human genome project to the present moment of AI and the life sciences: while the full potential of AI in biomedical research is not fully known yet, the field could look remarkably different a generation from now.
Launched in 2021, the Schmidt Center is enabling a new field of research at the intersection of machine learning and biology, aimed at improving human health. The Center's inaugural symposium, held on April 30 and May 1 at the Koch Institute For Integrative Cancer Research at MIT, brought together researchers and industry leaders who are advancing the foundations of machine learning and using these tools to understand the programs of life across scales, from proteins to cells to tissues and organisms.
The Schmidt Center inaugural symposium -- two exciting days of discovery, collaboration, and inspiration.
More than 250 in-person and 400 virtual attendees joined 27 speakers and panelists from across the Broad, MIT, Harvard, and beyond to learn how machine learning is uncovering insights into today’s most pressing biological questions, and in turn, how these questions are inspiring new directions in AI.
“We strongly believe that the biomedical sciences are not only well-positioned to benefit from machine learning, but they also offer some of the most exciting inspiration for foundational advances in ML,” said Caroline Uhler, Director of the Schmidt Center and Andrew and Erna Viterbi Professor of Engineering at MIT.
At the start of the symposium, Uhler noted that there had already been great energy – “I’m really looking forward to two exciting days of stimulating research and discussions,” she said.
Exploring Biology Across Scales
The symposium included a wide range of panels, talks, and poster presentations. The talks represented the breadth of the field, including using imaging and AI to understand the spatial organization of a tissue, molecular dynamics and functions at the subcellular level, and active drug discovery.
A common theme throughout the symposium was that science is at a pivotal moment, where the AI revolution will undoubtedly transform how researchers study the fundamental laws of life. Several discussions centered on whether and how a holistic foundation model for biology across scales, similar to ChatGPT in the language domain, could eventually be created, and what biological insights could be gained in the meantime from available data and simpler, more cost-effective machine learning frameworks.
The two panel discussions that concluded each of the days were complementary: one showcased experimentalists working on large-scale biological data generation, and the other offered insights from computational experts on foundation models across all scales (molecules, cells, tissues, organisms).
The symposium brought together well-established research leaders with researchers in the early stages of their independent careers, postdoctoral fellows, and PhD students.
Speaker Spotlights
The 11 invited talks featured Jennifer Lippincott-Schwartz (HHMI), Jean-Philippe Vert (Owkin, Bioptimus), GV Shivashankar (ETH Zurich), Susanne Rafelski (Allen Institute for Cell Science), Dana Pe’er (Memorial Sloan Kettering Cancer Center), Richard Bonneau (Genentech), Emma Lundberg (Stanford), Maria Brbic (EPFL), Anshul Kundaje (Stanford), Eric Xing (GenBio AI, CMU, MBZUAI), and Patrick Hsu (Arc Institute, UC Berkeley).
During the panels, discussions were led by Shantanu Singh (Broad), Xiaowei Zhuang (Harvard, HHMI), Fei Chen (Broad), Mark Daly (Broad, HMS, MGH), Sergey Ovchinnikov (MIT), Faisal Mahmood (HMS), Shirley Liu (GV20 Therapeutics), and Marzyeh Ghassemi (MIT), moderated by Eric Lander (Broad, MIT) and Caroline Uhler (Broad, MIT).
Joining them were 10 early career researchers – Athanasios Litsios (University of Toronto), Bo Xia (Broad, Harvard), Xinyi Zhang (Broad, MIT), Yakir Reshef (BWH, Broad), Yichen Si (Broad), Joey Bose (University of Oxford), Sandeep Kambhampati (Harvard, Broad), Pinar Demetci (Broad), and Michelle M. Li (HMS).
Scientific Highlights
Jennifer Lippincott-Schwartz (HHMI)
On the first day, Jennifer Lippincott-Schwartz illustrated how cutting-edge imaging technologies, combined with AI, allows researchers to study molecular dynamics in live cells at sub-cellular resolution, opening new doors to understanding cell function at multiple scales.
Dana Pe’er (Memorial Sloan Kettering Cancer Center)
Dana Pe'er presented computational methods developed by her lab to extract insights from spatial imaging data, define tissue niches, and help map how tissue architectures relate to function in health and disease.
Eric Xing (GenBio AI, CMU, MBZUAI)
On the second day, Eric Xing presented the idea of AI-driven “digital organisms” (AIDO) for simulating all biological phenomena. He noted that this could potentially be achieved by integrating and optimizing several modality-specific foundation models that are currently being developed at GenBio.
Shirley Liu (GV20 Therapeutics)
During the last session of the symposium, a panel on Foundation models in biology: DNA, Protein, Cells, Tissues, Organisms, Shirley Liu shared how AI can help identify targets for personalized cancer immunotherapy. She started from the idea that the immune system naturally produces antibodies to fight cancer, and by analyzing tumor-infiltrating immune cells, scientists can identify the tumor antigens that could amplify the endogenous immune response.
Early-Career Voices and Poster Winners
The symposium also gave early-career researchers an opportunity not only to present their posters, but to also participate in flash talks on both days, helping to share their work more widely.
The posters were judged by a committee that included Lindsay Edwards (Relation Therapeutics), Jean-Philippe Vert (Owkin, Bioptimus), and Orr Ashenberg (Broad).
Left to right: Lindsay Edwards (Relation Therapeutics), Anurendra Kumar, Uthsav Chitra, Xinhe Zhang, Caroline Uhler (Schmidt Center, Broad Institute)
Three poster winners emerged:
Uthsav Chitra (first place) – Mapping the topography of spatial gene expression with interpretable deep learning(Uthsav Chitra, Brian J Arnold, Hirak Sarkar, Kohei Sanno, Cong Ma, Sereno Lopez-Darwin, Shu Dan, Fenna Krienen, Benjamin J. Raphael)
Anurendra Kumar (second place) – CellWHISPER: Inference of contact-mediated cell-sell signaling(Anurendra Kumar, Nicholas Zhang, Bhavay Aggarwal, Ahmet Coskun, Saurabh Sinha)
Xinhe Zhang - (third place) – An AI-Cyborg System for Adaptive Intelligent Modulation of Organoid Maturation(Ren Liu, Zhaolin Ren, Xinhe Zhang, Qiang Li, Wenbo Wang, Zuwan Lin, Richard T. Lee, Jie Ding, Na Li, Jia Liu).
The poster presentations reflected the innovative, interdisciplinary nature of the field – blending computational rigor with biological insight.
“The level of science in the last couple of days has been absolutely phenomenal,” said Edwards. “We chose poster winners on the basis of three things: 1) originality; 2) scientific quality; and 3) scientific communication.”
Edwards expanded on the need for strong science communication skills, which includes compelling storytelling, clear visuals, and confident presentation, adding that those who communicate well are more likely to be invited to speak, ultimately amplifying the impact of their work.
Conversations Lead to Collaborations
During hallways conversations in between sessions, attendees reconnected with old colleagues and forged new collaborations, creating a lively, dynamic environment for idea-sharing across generations of researchers.
“This symposium brought together a lot of different perspectives to illuminate my research,” said Yue Qin, postdoctoral fellow at the Schmidt Center. “Just talking to people during the coffee breaks and lunch chats helped me figure out what is the next area that I should look into.”
Another Schmidt Center postdoctoral fellow – Viki Schuster – shared similar sentiments. “It was great to talk to people, get interesting questions, and discuss my work from different perspectives,” she said.
What's Next
Looking forward, speakers emphasized the importance of a coordinated global effort to standardize and aggregate datasets that are both consistent and representative of a broad spectrum of diseases. Future research should prioritize gaining novel biological insights, which may sometimes be achieved with simpler models rather than complex and resource-intensive foundation models. Continued work is required to better understand what these models learn, identify and mitigate potential biases, and recognize their limitations.
Todd Golub (Broad Institute)
As the Schmidt Center continues to advance biomedical discoveries and expand the community at the intersection of biology and AI, it looks forward to hosting this symposium annually, and continuing these conversations and collaborations.
“There’s that level of potential to really disrupt and transform how we think about biomedical research in the setting of machine learning and AI,” said Golub. “What a remarkable moment to be in science when, over the next decade, we are all going to figure out what the new world of biology and medicine is going to look like.”
A protein located in the wrong part of a cell can contribute to several diseases, such as Alzheimer’s, cystic fibrosis, and cancer. But there are about 70,000 different proteins and protein variants in a single human cell, and since scientists can typically only test for a handful in one experiment, it is extremely costly and time-consuming to identify proteins’ locations manually.
A new generation of computational techniques seeks to streamline the process using machine-learning models that often leverage datasets containing thousands of proteins and their locations, measured across multiple cell lines. One of the largest such datasets is the Human Protein Atlas, which catalogs the subcellular behavior of over 13,000 proteins in more than 40 cell lines. But as enormous as it is, the Human Protein Atlas has only explored about 0.25 percent of all possible pairings of all proteins and cell lines within the database.
Now, researchers from MIT, Harvard University, and the Broad Institute of MIT and Harvard have developed a new computational approach that can efficiently explore the remaining uncharted space. Their method can predict the location of any protein in any human cell line, even when both protein and cell have never been tested before.
Their technique goes one step further than many AI-based methods by localizing a protein at the single-cell level, rather than as an averaged estimate across all the cells of a specific type. This single-cell localization could pinpoint a protein’s location in a specific cancer cell after treatment, for instance.
Co-lead authors Xinyi Zhang and Yitong Tseo; Yunhao Bai
The researchers combined a protein language model with a special type of computer vision model to capture rich details about a protein and cell. In the end, the user receives an image of a cell with a highlighted portion indicating the model’s prediction of where the protein is located. Since a protein’s localization is indicative of its functional status, this technique could help researchers and clinicians more efficiently diagnose diseases or identify drug targets, while also enabling biologists to better understand how complex biological processes are related to protein localization.
“You could do these protein-localization experiments on a computer without having to touch any lab bench, hopefully saving yourself months of effort. While you would still need to verify the prediction, this technique could act like an initial screening of what to test for experimentally,” says Yitong Tseo, a graduate student in MIT’s Computational and Systems Biology program and co-lead author of a paper on this research.
Tseo is joined on the paper by co-lead author Xinyi Zhang, a graduate student in the Department of Electrical Engineering and Computer Science (EECS) and the Eric and Wendy Schmidt Center at the Broad Institute; Yunhao Bai of the Broad Institute; and senior authors Fei Chen, an assistant professor at Harvard and a member of the Broad Institute, and Caroline Uhler, the Andrew and Erna Viterbi Professor of Engineering in EECS and the MIT Institute for Data, Systems, and Society (IDSS), who is also director of the Eric and Wendy Schmidt Center and a researcher at MIT’s Laboratory for Information and Decision Systems (LIDS). The research appears today in Nature Methods.
Collaborating models
Many existing protein prediction models can only make predictions based on the protein and cell data on which they were trained or are unable to pinpoint a protein’s location within a single cell.
To overcome these limitations, the researchers created a two-part method for prediction of unseen proteins’ subcellular location, called PUPS.
The first part utilizes a protein sequence model to capture the localization-determining properties of a protein and its 3D structure based on the chain of amino acids that forms it.
The second part incorporates an image inpainting model, which is designed to fill in missing parts of an image. This computer vision model looks at three stained images of a cell to gather information about the state of that cell, such as its type, individual features, and whether it is under stress.
PUPS joins the representations created by each model to predict where the protein is located within a single cell, using an image decoder to output a highlighted image that shows the predicted location.
“Different cells within a cell line exhibit different characteristics, and our model is able to understand that nuance,” Tseo says.
A user inputs the sequence of amino acids that form the protein and three cell stain images — one for the nucleus, one for the microtubules, and one for the endoplasmic reticulum. Then PUPS does the rest.
A deeper understanding
The researchers employed a few tricks during the training process to teach PUPS how to combine information from each model in such a way that it can make an educated guess on the protein’s location, even if it hasn’t seen that protein before.
For instance, they assign the model a secondary task during training: to explicitly name the compartment of localization, like the cell nucleus. This is done alongside the primary inpainting task to help the model learn more effectively.
A good analogy might be a teacher who asks their students to draw all the parts of a flower in addition to writing their names. This extra step was found to help the model improve its general understanding of the possible cell compartments.
In addition, the fact that PUPS is trained on proteins and cell lines at the same time helps it develop a deeper understanding of where in a cell image proteins tend to localize.
PUPS can even understand, on its own, how different parts of a protein’s sequence contribute separately to its overall localization.
“Most other methods usually require you to have a stain of the protein first, so you’ve already seen it in your training data. Our approach is unique in that it can generalize across proteins and cell lines at the same time,” Zhang says.
Because PUPS can generalize to unseen proteins, it can capture changes in localization driven by unique protein mutations that aren’t included in the Human Protein Atlas.
The researchers verified that PUPS could predict the subcellular location of new proteins in unseen cell lines by conducting lab experiments and comparing the results. In addition, when compared to a baseline AI method, PUPS exhibited on average less prediction error across the proteins they tested.
Senior authors Caroline Uhler and Fei Chen
In the future, the researchers want to enhance PUPS so the model can understand protein-protein interactions and make localization predictions for multiple proteins within a cell. In the longer term, they want to enable PUPS to make predictions in terms of living human tissue, rather than cultured cells.
This research is funded by the Eric and Wendy Schmidt Center at the Broad Institute, the National Institutes of Health, the National Science Foundation, the Burroughs Welcome Fund, the Searle Scholars Foundation, the Harvard Stem Cell Institute, the Merkin Institute, the Office of Naval Research, and the Department of Energy.
Adapted from an article originally posted on MIT News.
As obesity continues to pose a major global health challenge, millions of people turn to GLP-1-based drugs, like Ozempic, for weight loss. But questions remain about who benefits most from these treatments. While prior research has examined demographic and lifestyle factors, the role of genetic variation in shaping individual responses has been largely unexplored.
In a new study published in Nature Medicine, researchers led by Jakob German, a PhD Fellow at the Eric and Wendy Schmidt Center, analyzed data from more than 10,000 individuals to investigate whether genetic risk influences how well people respond to two leading weight-loss interventions: GLP1-RAs and bariatric surgery. Working with collaborators from more than 40 institutions across six countries, the team combined genetic and clinical data from nine large-scale biobanks spanning Europe, North America, and the Middle East. By integrating clinical records and genomic profiles at this scale, they conducted one of the largest multi-ancestry studies to date examining how genetic differences affect responses to weight-loss treatments.
The researchers tested whether plausible genetic risk factors, including polygenic scores (PGS) for body mass index (BMI) and type 2 diabetes, could explain variability in weight loss after treatment. They found that genetic risk played little to no role in shaping weight loss for patients on GLP1-RA therapy. This suggests that GLP-1 therapies may offer consistent weight-loss benefits regardless of a person’s genetic predisposition to obesity or diabetes, making them a promising tool for equitable obesity care across diverse populations.
Bariatric surgery results revealed a similar story. While the PGS for BMI did reach statistical significance in some analyses, the effects were small — too minor to matter in a clinical setting. Together, these findings indicate that common genetic variation explains little of the variability in weight loss following surgery.
“Large-scale studies allow us to evaluate the real-world impact of genetics on treatment response,” said German. “Our results reinforce the idea that GLP1-RAs are broadly effective, independent of genetic background. It’s an encouraging finding for equitable and scalable obesity treatment.”
A key strength of the study was its integration of genomic and clinical data from diverse populations, underscoring the importance of scale in testing assumptions and guiding precision medicine. This approach aligns with the Schmidt Center’s mission to harness data and interdisciplinary science for transformative biomedical research.
The authors suggest future work could explore more targeted genetic predictors or examine individuals using GLP1-RAs specifically for obesity rather than type 2 diabetes. For now, their findings provide strong evidence that genetic risk alone should not limit access to GLP-1-based therapies and that these drugs may benefit many, regardless of inherited risk. Ultimately, studies like this help lay the foundation for more precise, data-driven approaches to understanding individual variation in response to GLP1-RAs and improving obesity care.
A new AI model developed by researchers at the Eric and Wendy Schmidt Center at the Broad Institute and ETH Zurich’s Department of Health Science and Technology can identify genes that have been altered, such as ones that might be causing a disease, in a cell just by analyzing an image of the cell’s chromatin — the dense package of chromosomes inside the cell’s nucleus. The machine learning tool, called Image2Reg, promises to accelerate both research on the genetic causes of disease and drug discovery by predicting drug targets and mechanisms.
In a study published recently in Cell Systems, a team led by longtime collaborators Caroline Uhler of the Schmidt Center and GV Shivashankar of ETH Zurich describe how their model can make predictions of genetic perturbations it has never encountered before. The scientists say that by bridging the gap between imaging data and molecular biology, Image2Reg offers a simple, rapid, and inexpensive alternative to more traditional sequencing-based approaches for mapping how cells respond to genetic or chemical changes.
Principal investigators and longtime collaborators Caroline Uhler and GV Shivashankar (photo credits: Schmidt Center; Mechanobiology Institute, NUS, Singapore/Melanie Lee)
“This is a prime example of what we aim to do at the Schmidt Center,” said Uhler, who is director of the Schmidt Center and the Andrew and Erna Viterbi Professor of Engineering in the Department of Electrical Engineering and Computer Science and Institute for Data, Systems, and Society at MIT. “By combining machine learning with biology at scale, we can reveal new layers of information from data that’s already being collected — and use it to guide therapeutic development.”
“As we continue to explore the potential of chromatin imaging, we’re seeing that it can serve as a powerful window into the regulatory state of the cell,” said Shivashankar, full professor at ETH Zurich and head of the Laboratory of Nanoscale Biology at the Paul Scherrer Institute.
How Image2Reg works
Disruptions in gene networks can lead to many diseases such as cancer and neurodegeneration. Uhler and Shivashankar have long been interested in the structure of chromatin because it can influence how these gene networks are regulated and contribute to disease. Fortunately for scientists, they have a rapid and inexpensive way to study chromatin: using fluorescent dyes to stain the chromatin and microscopes to capture images of it.
Inpreviouswork, Uhler and Shivashankar have shown that simple chromatin staining images combined with machine learning algorithms can yield a lot of information about the state and fate of a cell in health and disease. While machine learning has helped identify cell states, researchers hadn’t been able to trace them back to specific genes or regulatory programs.
To make these connections, the research team designed Image2Reg to learn from two types of data: chromatin images of cells with known genetic or chemical perturbations, and molecular profiles of gene interactions. First, the model uses a convolutional neural network to learn how different perturbations change chromatin structure. Then, it uses a graph-based model to learn how genes relate to each other in a specific cell type, using transcriptomics and protein-protein interaction data. Finally, a third component aligns these two embeddings, effectively translating between the physical organization of DNA and its biochemical regulation.
By successfully aligning chromatin structure with gene regulatory function, Image2Reg confirms a strong, predictive link between how DNA is physically organized and how genes behave — a connection that could help explain how diseases take hold at the molecular level.
This alignment enables Image2Reg to infer which genes are perturbed in new images it has never seen before. “By learning to map between representations of cell images and genes, our model can generalize to unseen perturbations, and that’s what makes it so powerful,” said Adityanarayanan Radhakrishnan, co-first author of the new study and postdoctoral fellow at the Schmidt Center.
Predicting drug effects
Co-first authors Daniel Paysan and Adityanarayanan Radhakrishnan; second author Xinyi Zhang (photo credits: Daniel Paysan; Schmidt Center)
To test Image2Reg’s ability to generalize, the team trained it on chromatin images of cells that each had one gene turned off or turned up to a high level. These image datasets — such as Cell Painting data from the Carpenter-Singh lab and perturbation screens from the Broad’s Cancer Dependency Map and JUMP-Cell Painting Consortium efforts — were generated at the Broad Institute and provided a rich foundation for training and validating the model. The model was able to predict the genetic targets of these drugs with 60 percent accuracy, even when it had never seen those compounds before.
“These results show that chromatin images can reveal how a compound affects the cell,” said Daniel Paysan, a co-first author, postdoctoral fellow at Novartis, and former Schmidt Center visiting PhD student. “We’re essentially using imaging to understand which genes a drug is targeting.”
While this study focused on a single cell type and specific perturbation conditions, the researchers say the approach is broadly applicable. As large-scale optical perturbation screens become more common, Image2Reg could be adapted to other experimental contexts — enabling scientists to study how gene regulation shifts in different cell types, disease states, or treatment responses.
Ultimately, the team hopes Image2Reg will become a foundation for linking chromatin structure to gene function at scale — helping researchers uncover the molecular mechanisms underlying disease and identify the genes that could be most effective to target with new or existing treatments.
Funding: This work was supported in part by the Swiss National Foundation, the Eric and Wendy Schmidt Center at the Broad Institute, the National Institutes of Health, the Office of Naval Research, AstraZeneca, the MIT-IBM Watson AI Lab, MIT J-Clinic for Machine Learning and Health, and a Simons Investigator Award.
The Eric and Wendy Schmidt Center, under the direction of Caroline Uhler, a core MIT IDSS faculty member, has convened investigators with machine learning and biomedical backgrounds from the Broad Institute, MIT, and Harvard to apply cutting-edge machine learning techniques to research focused on important challenges in biomedical science.
“These projects bring together scientists from interdisciplinary fields to generate large-scale datasets and develop new computational and algorithmic methods,” says Uhler. “This way, we can foster a two-way street between biology and machine learning, two disciplines that have so far largely developed in parallel, which holds the potential to create a new era of biology, yielding a deeper understanding of basic biological processes and improvements in human health.”
IDSS faculty members including Munther Dahleh, Philippe Rigollet, and Devavrat Shah, and their teams are involved in several of these projects, which are organized under research flagships that focus on critical questions in biomedicine. These research foci target areas where large-scale datasets and new computational methods can help catalog cell activity, identify causal relationships, and lead to a deeper understanding of underlying biological processes – insights which could cure disease and save lives.
The first flagship, titled ‘From genes to cell states: controlling cellular programs,’ focuses on the complex regulatory networks among genes that create the wide variety of cell types and functions. Recent technological advances in genomic perturbation, paired with new computational paradigms, could open a unique window into cell states and cell state transitions. One key aspect of this research explores causal relationships between cells and cell networks.
Projects under this flagship include research into determining cell intrinsic factors to induce desired cell state changes. Devavrat Shah’s group is contributing to define the transcription factor combinations that, when perturbed in human adipose-derived mesenchymal stem cells (AMSCs) ex vivo, give rise to the adipocyte subtypes observed in vivo. These projects will generate a multi-modal catalog of cellular programs to gain an understanding of the paths that can be taken by cells to move between AMSCs and different adipocyte subtypes.
Another area of research under this flagship focuses on determining cell intrinsic factors to induce desired cell state changes. Groups including Munther Dahleh’s are working toward mapping the full regulatory landscape of CD8 T cell subtypes/states. Their goal is to identify the optimal combination of genetic perturbations to obtain the T cell population ratio needed for the resolution of different diseases, with an initial focus on enhancing cancer immunotherapy.
“We are working on developing strategies for genetic perturbation to transform cells from one type to another,” explains Dahleh. “This is a highly complex problem due to the vast number of possible perturbations and the high cost of each experiment.” Dahleh brings his expertise in decision theory and intervention modeling to address this challenge through active learning, suggesting targeted interventions based on existing data and models to maximize information gain.
The second flagship organized by the Schmidt Center, titled “From cells to tissues: controlling cellular networks,” focuses on developing a multimodal foundation model of cells in tissues, where attention between cells can be interpreted causally to map out regulatory networks of cells in tissues.
Research under this flagship includes efforts to determine cell intrinsic and extrinsic programs underlying tissue zonation. Philippe Rigollet’s group joins efforts to map the cell intrinsic and extrinsic programs defining tissue zonation in the liver and the onset of tissue-restricted diseases such as liver steatosis, which are difficult to model in vitro.
“There are so many opportunities to be unlocked through collaborations between biological scientists, medical experts, and machine learning practitioners,” says IDSS director Fotini Christia. “Under Caroline’s leadership, the Schmidt Center has formulated a framework for advancing our understanding not only of genes, cells, tissues, and organs, but of disease and treatment as well. I am excited to see where this research goes, and how IDSS will contribute to it.”
As spring unfolds and our 2025 Models, Inference & Algorithms (MIA) season gains momentum, we invite you to explore our upcoming April and May talks. Whether you're following along in real time or catching up during spring break, we encourage you to review our playlist of past MIA talks, which we will continue to update. In the meantime, we have compiled a recap of the fall 2024 MIA meetings, complete with links to each talk. Happy watching!
September
Vidhi Lalchand (Schmidt Center) on "Bayesian optimization and its uses in the latent space of generative models"
1 - We started our fall MIA season with lightning talksfrom Ph.D. students and postdoctoral fellows across the Schmidt Center (Vidhi Lalchand), Harvard (Randy Ellis), MIT (Bowen Jing, Hannes Stark, Andreas Luttens), and the Whitehead Institute (Henry Kilgore). Their presentations covered a broad spectrum of machine learning applications, including protein subcellular compartmentalization, DNA sequence and protein ensemble generation, and dementia prediction using human biobank data.
October
Brian Cleary (Boston University). Photo: Boston University
2 -Brian Clearyand Aedan Brown, a Ph.D. student in Cleary’s lab, explored evolutionary dynamics in complex genotype-to-phenotype relationships. Using metabolism as a model, they showed that selection at the genetic level can be difficult to pinpoint, while selection on collective modes—preferred directions in phenotype space—provides a clearer picture of evolutionary dynamics and polygenic trait variability.
5 - Neriman Tokcanpresented tensor methods as powerful tools for analyzing high-dimensional multi-omics data. By extending traditional matrices into multi-way arrays, tensor methods capture complex interactions among genomic variables such as genes, samples, conditions, and omics layers. She also introduced Consensus-Zero Inflated Poisson Tensor Factorization (C-ZIPTF), a method developed by her lab to improve the stability of tensor factorization for zero-inflated genomic data – a common phenomenon in genomics.
November
Marnix Medema (Wageningen University)
6 - Marnix Medemapresented the ongoing efforts in his lab and the broader community to use computational and AI-driven methods to map the biosynthetic diversity of microbiome-derived metabolites and uncover their roles in microbe-microbe and host-microbe interactions. On the same topic, Victoria Pascal introduced gutSMASH, a tool for systematically analyzing microbial genomes for known and putative specialized primary metabolic gene clusters, and BiG-MAP, an algorithm for assessing gene cluster abundance and expression by using metagenomic and metatranscriptomic data as input.
Abraham Gihawi (University of East Anglia). Photo: University of East Anglia
7 - We concluded the fall season with a presentation by Abraham Gihawi and Steven Salzberg (videos not available), on cancer metagenomics. They explored the challenges of detecting microbiome DNA within tumor samples, addressing issues such as misclassified host sequencing reads and batch correction limitations.
Thank you for supporting the MIA community. We hope this recap makes it easy to revisit the highlights from fall 2024 and we look forward to seeing you at our future events!
Systems biology represents a powerful approach for revealing the molecular networks at play in model organisms through iterative cycles of perturbation, measurement, and analysis. However, applying this approach to human systems is inexact, and breaks a traditionally closed loop by introducing model systems that may not accurately capture the spatial and temporal complexity of human biology.
Former Schmidt Center postodctoral fellow David Fischer
David Fischer, former Eric and Wendy Schmidt Center postdoctoral fellow (2022–2024) and now an assistant professor at the Medical University of Vienna, leads a new perspective paper in Nature Reviews Genetics. Co-authored with Martin Villanueva (MIT) and Schmidt Center collaborators Peter Winter and Alex Shalek (Broad Institute), the paper presents a conceptual framework for studying human systems that accounts for the complexity of biological scales and helps bridge the "translational distance" between discoveries in human cohorts and model-based experimental validation.
Adapted from an update written by Broad Communications.
Traditionally, models of neural circuit development have proposed two phases: genetically driven neural wiring, followed by environmentally driven pruning and refinement. But this is inconsistent with certain innate behaviors; many animals, for example, are capable of complex problem solving just after birth.
Schmidt Center posdoctoral fellow Dániel Barabási.
In a perspective piece, Eric and Wendy Schmidt Center postdoctoral fellow Dániel Barabási, along with André Ferreira Castro (University of Cambridge) and Florian Engert (Harvard), propose a new three-system model for neural circuit formation. In the first system, development patterns neural circuits, defining innate understanding. In the second, “Eureka” learning events rapidly update existing knowledge. In the third, synapses are perpetually fine-tuned.
Recent advances in barcoding technologies have made it possible to reconstruct a lineage tree of cells while simultaneously capturing their transcriptomic profiles. However, to fully leverage the resolution provided by these lineage-resolved single-cell RNA sequencing (scRNA-seq) datasets, new computational approaches are needed. These methods must address key challenges, such as ensuring that gene expression analysis goes beyond pairwise comparisons between stages and instead captures the full hierarchical structure of lineage trees, allowing for the detection of gene expression patterns that follow or deviate from lineage relationships.
In a new study published in Nature Communications, researchers at the Eric and Wendy Schmidt Center at the Broad Institute of MIT and Harvard introduce PORCELAN, a statistical framework that automatically detects gene expression patterns linked to lineage progression. This method provides a systematic way to study how gene expression and cell state memory evolve through cell divisions, offering new insights into processes such as cancer progression.
Decoding Gene Expression Through Lineage Trees
PORCELAN – short for Permutation, Optimization, and Representation learning-based single Cell gene Expression and Lineage ANalysis – combines representation learning with permutations among leaves in the lineage tree. Using a statistical approach, PORCELAN addresses three questions: How can we jointly capture lineage and gene expression information in cell representations? Which genes best reflect lineage relationships, and in which subtrees is this connection strongest? To what extent does gene expression preserve lineage tree structure across different resolutions?
Schmidt Center graduate student and first author Hannah Schlueter aimed to create a rigorous and adaptable tool for studying cellular identity.
The researchers validated PORCELAN using synthetic datasets and applied it to three biological systems with lineage-traced scRNA-seq data: lung cancer progression, mouse embryogenesis, and C. elegans development. In lung cancer, PORCELAN identified tumor cell subpopulations that contributed to metastases and pinpointed key genes associated with these transitions – many of which align with known cancer biomarkers and pathways. In developmental systems, the framework uncovered differences in how gene expression memory is maintained across cell divisions, highlighting contrasts between normal development and cancerous progression. These findings underscore the importance of lineage-resolved approaches in understanding fundamental biological processes.
“Our goal was to develop a method that is both rigorous and adaptable,” says Schlueter. “Because PORCELAN is modular, it can be applied to different data modalities, including lineage-resolved imaging data, by replacing the simpler tree-likeness score based on local autocorrelation, used for transcriptomic data, with a representation learning-based tree-likeness score. This flexibility makes it a powerful tool for studying how cellular identity is maintained and altered over time.”
As lineage tracing technologies continue to evolve, methods like PORCELAN highlight the critical role of applying statistical techniques to biological research. This approach, which merges computational tools with biological insights, is central to the work at the Schmidt Center. By developing methods that bridge computational models with biological questions, the Schmidt Center aims to drive discoveries that deepen our understanding of cellular biology, disease mechanisms, and potential therapeutic strategies.
In a new study published this week by Nature, researchers at the Broad Institute of MIT and Harvard and Harvard University developed a new computational method – PRINT – that identifies DNA-protein interaction footprints from both bulk and single-cell chromatin accessibility data. By applying PRINT, researchers uncovered the organization and dynamics of cis-regulatory elements (CREs) across different scales, providing deeper insights into how genes are regulated, and paving the way for innovative research in both health and disease.
Advancing Gene Regulation Studies
CREs play a critical role in gene expression by binding to regulatory proteins, such as transcription factors (TFs) and histones, influencing fundamental biological processes. However, current methods for studying CREs at high resolution often rely on bulk data, which can mask the variability and specificity of regulatory elements in individual cells.
In this study, PRINT changes the landscape by analyzing both bulk and single-cell chromatin accessibility data to capture CRE organization at a high resolution. By combining PRINT with a deep learning framework, seq2PRINT, the authors were able to identify the binding dynamics of regulatory proteins across various cell contexts and states, such as cell differentiation and aging. This nuanced view enabled the authors to realize that CREs do not have only two regulation states (open/active or closed/inactive), but that the same CREs can be bound by different sets of TFs across cell types.
By enabling researchers to track changes in gene regulation in rare cell types or during disease progression, PRINT sheds light into gene regulation dynamics at the single-cell level in physiological and pathological conditions.
Yan Hu, graduate student at the Buenrostro Lab (image credit: Buenrostro Lab)
“Our study really helped open new opportunities to study how different TFs and nucleosomes combinatorially encode the regulation of gene expression, as well as nominating candidate factors driving diseases,” said co-first author Yan Hu, a graduate student at the Buenrostro Lab at Harvard.
The Buenrostro Lab aims to create sequencing technologies to better understand gene regulation across health and disease.
Ruochi Zhang, postdoctoral fellow at the Schmidt Center
The lab’s research heavily aligns with the Schmidt Center’s work, which converges biology and machine learning to drive biological discoveries. By blending advanced computational methods with experimental biology, PRINT represents the kind of innovation that emerges from interdisciplinary collaboration.
“This is a breakthrough that we couldn’t have accomplished alone without bridging biology and AI through this collaboration,” said Zhang. “Biology and AI form a two-way street—the diverse expertise within our team provides different perspectives on the problem, motivates innovative approaches for investigation, and ultimately drives deeper understanding of the questions we’re addressing.”
Max Horlbeck, geneticist at Boston Children’s Hospital and postdoctoral fellow at the Buenrostro Lab (image credit: Max Horlbeck)
Hu, Horlbeck, and Zhang co-led the efforts of the research, with contributions from colleagues at the Buenrostro Lab, Wagers Lab, and the Gene Regulation Observatory (GRO) at the Broad Institute. This work was supported by NIH institutes NHLBI, NHGRI, NIGMS, NICHD, and the common fund, Broad Institute, Schmidt Center, the GRO, Wagers Lab, Harvard Department of Stem Cell and Regenerative Biology, Harvard Stem Cell Institute, and the Impact of Genomic Variation on Function Consortium. Read more about their work in Nature.
As dynamic as the systems that he researches, Matt Levine brings his friendliness, mathematical knowledge, and eagerness to collaborate and help others to the Schmidt Center.
After studying biophysics at Columbia University, the New Jersey native found his passion for computational projects as opposed to lab work. He joined a research project on diabetes with David Albers, George Hripcsak, and Lena Mamykina, quickly realizing that he enjoyed tackling complex dynamics problems.
Matt also collaborated with mathematician and his future PhD advisor Andrew Stuart, who showed him the clarifying power of math and encouraged him to pursue a PhD in applied mathematics at Caltech. As Matt figured out that he wanted to return to some of his biological roots, he worked with Michael Elowitz on biological computations during his final year of graduate school, and decided to pursue a fellowship at the intersection of applied math, machine learning, statistics, and biomedicine, leading him to the Schmidt Center in the fall of 2023.
In his research on dynamical systems, Matt finds a balance between combining traditional, physics-based models with modern AI techniques. “It’s easy for mathematicians to work on theoretical models that seem perfect on paper, but fail in practice,” he says. “This is why we need to ground ourselves in real data and questions to ensure that the theories work in real-world scenarios.”
Read on to learn more about Matt’s numerous collaborations, this year’s conference presentations, and travel highlights.
Matt at his Caltech graduation.
Tell us about your area of research.
A lot of my research centers around dynamical systems, which are systems that evolve over time. My focus is on understanding these systems by learning from the data we collect, which often involves developing models to explain how these systems work or to predict their future behavior.
I think about this problem by blending old-school, knowledge-based approaches with newer techniques. The idea is that we often have a solid understanding of a system before we even start collecting data. Rather than relying solely on modern AI approaches that learn everything from scratch, I believe in using a hybrid style. This means we start with what we know and use that knowledge to fill in the gaps where we don’t have information.
I apply these principles in an application-agnostic setting, or a broad way where the methods are applicable to many different types of problems. However, I also focus on specific applications to make sure that the rubber can meet the road. I test these models on actual problems, observe where they fall short, and analyze why they didn’t work. After I get things to work in that setting, it’ll eventually fail again, creating a loop, and that’s the loop I like to live in.
A lot of the applications that I've worked on have a biomedical focus. For example, I’ve worked on modeling glucose dynamics in people with diabetes, using real-world data from daily self-monitoring to gain insights into their physiology. I’ve also applied these methods to climate science, aiming to improve climate models by using data to calibrate and refine these models.
What’s something you wish more people understood in regards to your research?
I wish that more people would recognize the power of mathematics. It’s a powerful language that can be used as a clear, precise, and efficient communication tool. Even when working with experimentalists, writing down the plan and evaluations in terms of rigorous mathematics is a clarifying exercise. By getting it into such a clear language, math brings up questions that wouldn’t have been asked otherwise, forcing you to look at new things carefully.
Matt has worked substantially in the biomedical sciences, and enjoys collaborating on impactful applied projects.
Tell us about one of your collaborations.
I’m excited about a new collaboration with Luca Pinello, who’s exploring cell fate and differentiation as dynamic processes. He’s interested in how cells evolve over time, but instead of watching a continuous "video" of a cell’s entire lifespan, we work with snapshots—random moments in the cell's life. It’s like taking a snapshot of Earth today and trying to understand aging; you get different snapshots at different times but can't connect one person’s past and future.
This challenge raises interesting methodological questions and has led to discussions on various dynamical systems and modeling approaches. Our collaboration has led to new mathematical definitions and innovative methods. I'm excited to continue learning from Luca’s group, while sharing insights from my work – it’s been rewarding working together.
You presented at several major conferences this year.
I went to two exciting conferences in July. The first was ICML (International Conference on Machine Learning) in Vienna, where I gave an oral presentation on a paper I have with a group from Stanford (Emily Fox, Ramesh Johari, Dessi Zaharieva, Bob Junyi Zou) – Hybrid Neural ODE Causal Modeling and an Application to Glycemic Response. We focused on learning dynamics from data collected from people with diabetes. Because the data was noisy and sparse, we had to use a lot of existing knowledge to improve our methods, which led us to develop some new approaches.
Before and after the conference, I also spent time in upper Austria on the weekends with a friend from grad school. It was really peaceful – we hiked, swam in lakes, observed the scenery, watched the cows, ate the sausage, and had a great time.
At the Fourth Symposium on Machine Learning and Dynamical Systems at the Fields Institute in Toronto, I presented work done in collaboration with a group from Caltech (Andrew Stuart, Edoardo Calvello, Nikola Kovachki) during my PhD. We’re exploring how large language models (LLMs) and their Transformer architectures, typically used for sequences like word lists, can be adapted for continuous data such as time series or images. Our new approach, called operator learning, reformulates these models to handle data of varying resolutions. This allows our models to process and understand data consistently, even when resolution changes, like in pathological images with different pixel densities.
Matt's never bored as long as he has a board to jot down his ideas.
What’s a memorable experience you had related to your research?
I recently had the opportunity to collaborate with a friend – Iñigo Urteaga – from my Columbia days. Although we weren’t in the same research group, we were close friends and colleagues—he was a postdoc while I was still a pre-doc, before I applied to graduate school.
In the summer of 2023, I visited Iñigo at the Basque Center for Applied Mathematics in Bilbao, Spain. With him starting a new professorship there and me about to begin a postdoc at Broad in the fall of 2023, it was the ideal moment for us to reconnect and tackle a project we had long discussed. Our collaboration focuses on uncertainty quantification in dynamical systems—a field that examines how we can model systems from data and gauge the range of possible models that could explain our observations. This approach helps us understand not just what the data tells us, but how confident we should be about different explanations. We developed a JAX package that’s available on GitHub.
I visited him again last winter, and he’s been a formal visitor at MIT and Broad since October. We’re continuing to work together and he’s had a chance to meet the Schmidt Center fellows and start other collaborations.
Mostly, it was really nice to work in Bilbao. We’d take coffee breaks in cafes, drink our espresso, then have long, Spanish lunches. It was a pleasant, peaceful environment to get work done, making our collaboration particularly rewarding.
"It's very cathartic for me to spend time in the mountains, with the mountain air, in the middle of nowhere," says Matt.
Let’s talk about the Oberwolfach Research Institute for Mathematics (MFO).
Coined “Math Camp” by a friend and located in the Black Forest, Germany, the MFO conference program hosts 20-30 researchers for focused seminars and workshops. When I attended, the schedule was relaxed, with lectures, coffee breaks, and group lunches, and every Wednesday we would hike and enjoy Black Forest cake together— it’s a whole tradition.
During one of these hikes, I was talking to a researcher from Youssef Marzouk’s group about challenges I faced with learning differential equations from partially observed data. That conversation sparked an idea about using data assimilation, which turned out to be a breakthrough. I went back to my room, ran some code, and the next day I had a working solution for something I'd been struggling with for over a year during my PhD. I was very excited and shared my findings with the group in an impromptu talk, and this idea eventually contributed to a paper I was working on. I'm still exploring similar concepts today with Youssef’s group, focusing on uncertainty quantification for machine learning in unobserved systems.
What advice would you give to aspiring researchers?
One of the aspects I truly enjoy about academia is the emphasis on collaboration and knowledge sharing. I find a lot of joy in working with others and believe that a friendly, open environment greatly enhances the productivity and satisfaction of everyone involved.
When people ask how I’ve managed to build many collaborations, my answer is simple: being friendly goes a long way. I’ve noticed that many successful researchers share this trait. For instance, George Hripcsak, the former chair of the Department of Biomedical Informatics at Columbia, is a prime example. His success in leading large consortia and managing diverse projects largely comes from people enjoying working with him. He’s great at what he does and he’s approachable. In my experience, being kind, open, and personable often proves to be more effective than just being a research machine. Be friendly!
What are some of your hobbies?
I love skiing – it’s very cathartic for me to spend time in the mountains, with the mountain air, in the middle of nowhere. Last winter I was an adaptive ski instructor, which was a really rewarding experience, as I worked with people who had specific needs, and tailored their skiing lessons accordingly. Whether it involved using specialized equipment or teaching them one-on-one, the goal was to make skiing accessible and enjoyable for everyone. It was definitely the hardest job I ever had, but incredibly fulfilling. I also like playing tennis and music, specifically the piano and guitar. I enjoy doing activities because they help me be present in the moment in a physically engaging way, which is different from my regular job.
Nothing brings Matt down -- after he falls, he bounces back up, with a smile.
Some of Matt’s Favorite Things:
Underrated technology: Metronome
Game: Chess
Movie: Office Space
Book: A Visit from the Goon Squad by Jennifer Egan
Snack: Cadbury chocolate
Element on the periodic table: Ununennium
Anything else? I’m very appreciative of my mentors!
Each year at the Retreat, in recognition of their exceptional contributions to the institute, Broad presents outstanding Broadies with the Broad Institute Excellence and Achievement Awards as well as the Eric S. Lander and Stuart L. Schreiber Awards in Scientific Excellence. This year, the Eric and Wendy Schmidt Center is thrilled to announce that Schmidt Center PhD Fellow Jiaqi Zhang was among one of the winners.
The Eric S. Lander and Stuart L. Schreiber Awards in Scientific Excellence were established in 2019 to recognize outstanding scientists at the Broad who demonstrate, first and foremost, scientific excellence, along with exceptional commitment to promoting women in science — either by their own example or through mentoring and supporting women in their scientific careers. They demonstrate true Broadie spirit and show what makes the Broad so special. These rising stars presented their research during the award ceremony, and were recognized with a commemorative scientific excellence award and an accompanying $1,000 prize. They will also participate in a Broad-sponsored leadership development program and will serve on the award selection committee next year.
Jiaqi receiving her award at the Broad Retreat.
Jiaqi Zhang began her academic journey at Peking University, where she earned a bachelor’s degree in statistics. She is now a Ph.D. candidate in the Department of Electrical Engineering and Computer Science at MIT, a researcher in the Schmidt Center within the Uhler Lab at Broad, and co-leader of the Machine Learning subgroup in the Gene Regulation Observatory.
Jiaqi’s research focuses on causal inference and experimental design. She has developed active learning algorithms that enable efficient identification of optimal genetic interventions, which reduces experimental costs and increases precision. Her work has advanced fields such as cancer immunotherapy and cellular reprogramming and has led to numerous first-author publications in top journals. She has proven herself to be an emerging leader in machine learning and computational biology.
A cornerstone of Jiaqi’s contributions has been her work on a large-scale cancer immunotherapy data science challenge last year at the Broad, which attracted more than 1,000 participants globally. She co-designed and analyzed machine learning tasks to identify genetic targets that enhance T-cell efficacy against cancer. Jiaqi is currently in the final stages of preparing a first-author manuscript on the novel perturbations identified through this deep integration of computation and experimental work.
Beyond her scientific contributions, Jiaqi is a dedicated advocate for gender equity in science. She has consistently mentored undergraduate and early-stage graduate students. Her active participation in initiatives like the Women in Data Science Conference and the Rising Star in EECS workshop underscores her dedication to building a supportive and inclusive scientific community, inspiring more women to pursue and excel in STEM fields.
Adapted from an article written by Broad Communications.
With more than 10,000 nominations each year, the Under 30 list, evaluated by Forbes editors and industry expert leaders, recognizes a total of 600 young innovators under the age of 30 from North America, within 20 different industries, who are creating meaningful change with business, culture, and entrepreneurship. The science candidates were evaluated by a panel of judges featuring Sara Seager, professor of physics at MIT; Luna Yu, cofounder and CEO of Genecis; Christina Smolke, cofounder and CEO of Antheia; and Randy Glein, cofounder and partner of DFJ Growth.
Dániel joined the Schmidt Center as a fellow in 2024, collaborating with Professors Xiao Wang and Jason Buenrostro. He holds a PhD from the Harvard Biophysics Graduate Program, where he was advised by Florian Engert (2024), and a bachelor’s degree in physics from the University of Notre Dame, where he worked with Zoltán Toroczkai on distance-based network models of brain connectivity (2013-2017).
By characterizing brain connectivity maps and combining them with high-throughput transcriptomics, Dániel looks to uncover the architectural building blocks of neural circuits, thereby understanding the brain’s maturation, wiring, and function. In time, he aims to apply these tools to study evolutionary differences in brain connectivity between species and investigate how stress dysregulates the brain, with the goal of informing therapeutic interventions.
By studying changes in gene expression, researchers learn how cells function at a molecular level, which could help them understand the development of certain diseases.
But a human has about 20,000 genes that can affect each other in complex ways, so even knowing which groups of genes to target is an enormously complicated problem. Also, genes work together in modules that regulate each other.
Researchers from MIT and the Eric and Wendy Schmidt Center at the Broad Institute of MIT and Harvard have now developed theoretical foundations for methods that could identify the best way to aggregate genes into related groups so they can efficiently learn the underlying cause-and-effect relationships between many genes.
Importantly, this new method accomplishes this using only observational data. This means researchers don’t need to perform costly, and sometimes infeasible, interventional experiments to obtain the data needed to infer the underlying causal relationships.
In the long run, this technique could help scientists identify potential gene targets to induce certain behavior in a more accurate and efficient manner, potentially enabling them to develop precise treatments for patients.
Schmidt Center fellow and MIT EECS PhD student Jiaqi Zhang
“In genomics, it is very important to understand the mechanism underlying cell states. But cells have a multiscale structure, so the level of summarization is very important, too. If you figure out the right way to aggregate the observed data, the information you learn about the system should be more interpretable and useful,” says Schmidt Center Fellow Jiaqi Zhang, MIT graduate student in the Department of Electrical Engineering and Computer Science (EECS), and co-lead author of a paper on this technique.
Zhang is joined on the paper by co-lead author Ryan Welch, also a Schmidt Center fellow and MIT master’s student in engineering; and senior author and Schmidt Center Director Caroline Uhler, Andrew (1956) and Erna Viterbi Professor of Engineering in EECS and the Institute for Data, Systems, and Society (IDSS) at MIT, and a researcher at MIT’s Laboratory for Information and Decision Systems (LIDS). The research will be presented at the Conference on Neural Information Processing Systems (NeurIPS).
Schmidt Center fellow and MIT master's student Ryan Welch
Learning from observational data
The problem the researchers set out to tackle involves learning programs of genes. These programs describe which genes function together to regulate other genes in a biological process, such as cell development or differentiation.
Since scientists can’t efficiently study how all 20,000 genes interact, they use a technique called causal disentanglement to learn how to combine related groups of genes into a representation that allows them to efficiently explore cause-and-effect relationships.
In previous work, the researchers demonstrated how this could be done effectively in the presence of interventional data, which are data obtained by perturbing variables in the network.
But it is often expensive to conduct interventional experiments, and there are some scenarios where such experiments are either unethical or the technology is not good enough for the intervention to succeed.
With only observational data, researchers can’t compare genes before and after an intervention to learn how groups of genes function together.
“Most research in causal disentanglement assumes access to interventions, so it was unclear how much information you can disentangle with just observational data,” Zhang says.
The MIT researchers developed a more general approach that uses a machine-learning algorithm to effectively identify and aggregate groups of observed variables, e.g., genes, using only observational data.
They can use this technique to identify causal modules and reconstruct an accurate underlying representation of the cause-and-effect mechanism. “While this research was motivated by the problem of elucidating cellular programs, we first had to develop novel causal theory to understand what could and could not be learned from observational data. With this theory in hand, in future work we can apply our understanding to genetic data and identify gene modules as well as their regulatory relationships,” Uhler says.
Schmidt Center Director and MIT EECS and IDSS Professor Caroline Uhler
A layerwise representation
Using statistical techniques, the researchers can compute a mathematical function known as the variance for the Jacobian of each variable’s score. Causal variables that don’t affect any subsequent variables should have a variance of zero.
The researchers reconstruct the representation in a layer-by-layer structure, starting by removing the variables in the bottom layer that have a variance of zero. Then they work backward, layer-by-layer, removing the variables with zero variance to determine which variables, or groups of genes, are connected.
“Identifying the variances that are zero quickly becomes a combinatorial objective that is pretty hard to solve, so deriving an efficient algorithm that could solve it was a major challenge,” Zhang says.
In the end, their method outputs an abstracted representation of the observed data with layers of interconnected variables that accurately summarizes the underlying cause-and-effect structure.
Each variable represents an aggregated group of genes that function together, and the relationship between two variables represents how one group of genes regulates another. Their method effectively captures all the information used in determining each layer of variables.
After proving that their technique was theoretically sound, the researchers conducted simulations to show that the algorithm can efficiently disentangle meaningful causal representations using only observational data.
In the future, the researchers want to apply this technique in real-world genetics applications. They also want to explore how their method could provide additional insights in situations where some interventional data are available, or help scientists understand how to design effective genetic interventions. In the future, this method could help researchers more efficiently determine which genes function together in the same program, which could help identify drugs that could target those genes to treat certain diseases.
This research is funded, in part, by the MIT-IBM Watson AI Lab and the U.S. Office of Naval Research.
At first, Eric and Wendy Schmidt Center postdoctoral fellow Yue Qin thought she wanted to become a doctor. She’d always been interested in disease — why people got sick, why some illnesses could send you to a hospital while others could be treated at home. As she grew older, however, she realized she was more interested in learning about the roots of disease and the genes that caused them.
Growing up in Ningbo, a city in eastern China, Qin was strongly influenced by societal expectations that girls were better suited for language arts than math and science. She assumed that her struggles in a high school computer science course were because of her gender, even though she’d had no trouble with math. That all changed in college, at the University of California, San Diego (UCSD), when Qin took an introductory computer science course and fell in love with coding and its logic. She began seriously considering a career in computer science and combining it with her interest in biology.
Qin loves being at the intersection of computer science and biology because "it opens up new avenues of scientific exploration and pushes the limit of what we are able to do," she says.
Doing computational biology research as an undergraduate, as well as having supportive research advisors, inspired her to stay in research. After graduating with a bachelor’s degree in bioinformatics, Qin then went on to pursue her PhD at UCSD, where she used computational modeling to study how proteins interact with each other and assemble into a human cell. In January 2023, she joined the Schmidt Center as a postdoctoral fellow in the labs of Paul Blainey and Director Caroline Uhler. At the Broad, she aims to create an in silico cell: a computational model scientists can use to study at scale how external influences such as drug treatments affect cells.
We spoke with Qin about finding her place in science, how computational tools can advance biological research, and what it’s like to do both computational and wet lab experiments in this #WhyIScience Q&A.
What do you like about the intersection of computer science and biology?
What first intrigued me about computer science was that once you understand the logic of code, everything makes sense. You write a code and enter it into a computer and it will just do whatever the code says. Any errors are part of the code itself. Once I understood it, I found I was really in love with this logic. And in biology, you don't always understand what goes wrong in disease, how mistakes in our genome get translated. All those pieces are missing. But biology also has its own form of logic, and I was curious if I could use the logic in computer science to help me understand the logic in biological science.
Studying only biology limits you to a specific type of research, and the same goes for computer science. While the research within each discipline can be quite distinctive, when you merge these different fields, then you have amazing things like [the AI protein-structure prediction algorithm] AlphaFold. Before, biologists haven't been able to study the function of certain proteins because their structures were unknown. Now, with AlphaFold predictions, biologists have so many new hypotheses.
Being at the leading edge of science that intersects with computer science and biology is super exciting. It opens up new avenues of scientific exploration and pushes the limit of what we are able to do.
How are you using computational methods to study biological systems?
My goal is to build biotechnology tools and computational methods that can enable us to create an in silico cell to simulate interventions of treatments so that we can understand and treat disease.
Perturbing genes at scale in a dish is one of the easiest and most cost-effective ways to help us understand the functions of genes and find new therapeutic options. The problem we're really facing is that, for example, knocking out a single gene is not enough to cure cancer. We might need to treat with two drugs or perturb two pathways simultaneously to effectively cure cancer. We have 20,000 genes and if we want to exhaustively explore the effects of perturbing any two genes, that's 200 million options. But people have found that even perturbing just two pathways won’t be enough. The problem is just enormous; it's not solvable purely in the lab. However, by understanding how genes interact with each other using existing knowledge in an in silico cell, we can simulate unseen relationships between genes — even if we haven't seen this perturbation, models can learn from the existing data — to predict what we’d see in a dish.
With the help of machine learning, we really scale down the number of experiments that we have to do and can focus on the path that's most promising for therapeutics.
Qin with members of Caroline Uhler's lab.
What is it like to juggle computational and wet lab research?
In undergrad I took both biology and computer science courses. But what I found frustrating was that even with experience in both fields it was still hard to communicate to bench scientists because you need to get into really technical details and I didn’t know them. I thus decided to dive more into the wet lab so that I could bridge the two fields and drive effective collaborations. I decided that during my postdoc I wanted to get training in both.
What are you working on now?
In one project, I've been using cell images from Paul Blainey’s lab, where we perturb the genome using a new biotechnology tool that's currently on bioRxiv called CROPseq-multi, which allows us to look at genetic interactions in the image space in a pooled fashion. That's something that we could never do before because we just didn't have the tools. In the past, scientists have used images mainly in small-scale experiments to validate hypotheses, taking a few under the microscope to see if expected phenotypes show up. But we now finally have the power and the technology to easily generate large image datasets, empowering machine learning to help us understand what changes in cell morphology mean and connect morphology and genetics at a genome-wide scale.
How could an in silico cell improve the treatment of disease?
I wasn’t always aware of the imbalance in access to medical resources. My grandpa in Rizhao [a city in northeastern China] had cancer and even though I work in the field of cancer research, there was nothing I could do to help him. This made me realize that I'm in a privileged setting where I'm surrounded by medical experts, but the amount of medical resources that he had was totally incomparable to the ones I'm exposed to. That really got me thinking about how we could address such disparities, and one approach could be using an in silico cell to simulate different disease contexts using patient information including genomics and give therapeutic options to patients. With such a model, we could alleviate the inequitable access to medical knowledge for patients around the world.
We also need new therapeutics and there are so many diseases where we don’t know the cause and don’t even have a therapeutic option available for patients who are suffering. This research can help us find the direct pathway that we should target in personalized genetic contexts. Hopefully this can inspire new therapeutic developments from pharmaceutical companies.
Adapted from an article posted on the Broad site.
The Eric and Wendy Schmidt Center at the Broad Institute of MIT and Harvard is pleased to announce that Center Director Caroline Uhler has been named the Andrew (1956) and Erna Viterbi Professor of Engineering, effective July 1, 2024 for a five-year term. This MIT School of Engineering Professorship is awarded to an outstanding faculty member who is recognized as a leader and innovator in the field of Electrical Engineering and Computer Science.
Caroline holds BSc degrees in math and biology, an MSc in mathematics, and an MEd in mathematics education from the University of Zurich (years spanning 2004-7), and a PhD in statistics from UC Berkeley (2011). Before joining MIT as a faculty member in 2015, she spent three years as an assistant professor at IST Austria.
Caroline’s research focuses on machine learning methods for integrating and translating between vastly different data modalities and inferring causal or regulatory relationships from such data. She is particularly interested in using these methods to gain mechanistic insights into the link between genome packing and regulation in health and disease.
She is an elected member of the International Statistical Institute, and is the recipient of a Simons Investigator Award, a Sloan Research Fellowship, and an NSF Career Award. Recently, she was named a Fellow of the Institute of Mathematical Statistics (IMS), 2024, and a Fellow of the Society for Industrial and Applied Mathematics (SIAM), Class of 2023.
Ductal carcinoma in situ (DCIS), a pre-invasive tumor, accounts for about 25% of breast cancer diagnoses, a leading cause of cancer death. While doctors generally recommend treatment, they lack the appropriate evidence to reliably decide which tumor will remain benign and which might turn into a life-threatening invasive ductal carcinoma (IDC), resulting in high rates of overtreatment.
The current methods for understanding DCIS progression include manual assessment of nuclear morphology by pathologists, sequencing-based approaches, spatial transcriptomics, and highly multiplexed imaging. However, these methods face challenges due to cost, complexity, and limited information about the tissue microenvironment, which is necessary for accurate DCIS progression assessment.
In a new study published today in Nature Communications, researchers at the Broad Institute of MIT and Harvard and the Paul Scherrer Institute at ETH Zürich in Switzerland have found a simple and effective method of predicting the disease stage of DCIS, which could ultimately lead to more informed recommendations for DCIS breast cancer treatment. Their analysis demonstrates that, without the need of multiple stains or sequencing-based technologies, chromatin imaging provides sufficient information about cell states and tissue organization to accurately predict tumor stages.
Caroline Uhler says that using machine learning to analyze data can ultimately lead to better disease diagnosis and treatment.
Shivashankar’s lab is interested in understanding the underlying mechanisms for cell-state transitions and the association with disease states. They aim to improve early disease diagnostics by using multi-disciplinary approaches, such as single-cell imaging, functional genomics, and machine learning, to study the coupling between cell mechanics and genome organization in tissue contexts.
“Building on our previous studies with the Schmidt Center, we’re thrilled that we found a simple way to predict disease stage through the statistics of cell states, and we look forward to seeing how this can be applied to DCIS treatment,” said co-senior author Shivashankar.
G.V. Shivashankar is currently developing various methods for the diagnosis and prognosis of cancer at PSI (image credit: PSI).
The study aligns with the Schmidt Center’s goal of fostering a two-way street between biology and machine learning to advance biomedical discoveries and provide insights into how cells work in health and disease.
“As our research on DCIS shows, it’s important to create novel machine learning methods to analyze biomedical data,” said co-senior author Uhler. “Using machine learning to analyze data can lead to more accurate and simpler solutions for important biological questions, ultimately leading to better disease diagnosis and treatment.”
“Collaborating with Shivashankar’s lab, which I have done on several projects, provides me with a unique opportunity to develop computational methods for important biological problems,” said study first author Xinyi Zhang, a Ph.D. student at MIT and the Schmidt Center. “I’m able to see what the real roadblocks and challenges are in the biomedical space and start thinking about what to develop next.”
Xinyi Zhang has collaborated with Shivashankar’s lab multiple times, develop computational methods for important biological problems (image credit: Jared Charney).
Using unsupervised representation learning methods, the scientists analyzed 560 samples from 122 patients at 11 stages of DCIS progression from normal to cancerous breast tissues. They identified eight disease-relevant cell states based on nuclear morphology and chromatin organization, and found that all eight cell states exist in all disease stages, but with different abundances.
Based on the learned representations, the researchers then arranged the cell types from healthy to cancerous, finding that the order matched the natural progression of the disease, even though the model wasn't trained directly on disease stages. The study also demonstrated that spatial organization of cells near breast ducts and the co-localization of cell states can better predict disease stage compared to cell state abundance alone. This approach highlighted distinct cell states, their relative abundances, and their spatial neighborhoods, indicating their potential as biomarkers for cancer staging.
Although follow-up clinical trials with longitudinal tracking of DCIS patients are needed, this study demonstrated that high-dimensional AI-inferred features based on simple and cheap chromatin images can provide valuable insights into tumor progression. Uhler noted that this study introduces a new approach to exploring disease progression within a tumor microenvironment, specifically by leveraging machine learning and computational methods to extract meaningful information from complex chromatin images, without the need for extensive staining or sequencing.
By focusing on one of the Schmidt Center’s core missions – developing the foundations of machine learning to understand the programs of life – this study offers simple and cost-effective solutions for disease prognosis and treatment.
With recent advances in imaging, genomics and other technologies, the life sciences are awash in data. If a biologist is studying cells taken from the brain tissue of Alzheimer’s patients, for example, there could be any number of characteristics they want to investigate — a cell’s type, the genes it’s expressing, its location within the tissue, or more. However, while cells can now be probed experimentally using different kinds of measurements simultaneously, when it comes to analyzing the data, scientists usually can only work with one type of measurement at a time.
The fourth-year MIT PhD student is bridging machine learning and biology to understand fundamental biological principles, especially in areas where conventional methods have hit limitations. Working in the lab of MIT Professor and Schmidt Center Director Caroline Uhler in the Department of Electrical Engineering and Computer Science (EECS), the Laboratory for Information and Decision Systems (LIDS), and the Institute for Data, Systems, and Society (IDSS), and collaborating with researchers at the Schmidt Center, Zhang has led multiple efforts to build computational frameworks and principles for understanding the regulatory mechanisms of cells.
Xinyi says she wants to keep applying her skills to solve the “most challenging questions that we don’t have the tools to answer.”
“All of these are small steps toward the end goal of trying to answer how cells work, how tissues and organs work, why they have disease, and why they can sometimes be cured and sometimes not,” Zhang says.
The activities Zhang pursues in her down time are no less ambitious. The list of hobbies she has taken up at the Institute include sailing, skiing, ice skating, rock climbing, performing with MIT’s Concert Choir, and flying single-engine planes. (She earned her pilot’s license in November 2022.)
“I guess I like to go to places I’ve never been and do things I haven’t done before,” she says with signature understatement.
Uhler, her advisor, says that Zhang’s quiet humility leads to a surprise “in every conversation.”
“Every time, you learn something like, ‘Okay, so now she’s learning to fly,’” Uhler says. “It’s just amazing. Anything she does, she does for the right reasons. She wants to be good at the things she cares about, which I think is really exciting.”
Zhang first became interested in biology as a high school student in Hangzhou, China. She liked that her teachers couldn’t answer her questions in biology class, which led her to see it as the “most interesting” topic to study.
Her interest in biology eventually turned into an interest in bioengineering. After her parents, who were middle school teachers, suggested studying in the United States, she majored in the latter alongside electrical engineering and computer science as an undergraduate at the University of California at Berkeley.
Xinyi with other members of Caroline Uhler's lab.
Zhang was ready to dive straight into MIT’s EECS PhD program after graduating in 2020, but the COVID-19 pandemic delayed her first year. Despite that, in December 2022, she, Uhler, and two other co-authors published a paper in Nature Communications.
The groundwork for the paper was laid by Broad Institute core member Xiao Wang, one of the co-authors. She had previously done work with the Broad Institute in developing a form of spatial cell analysis that combined multiple forms of cell imaging and gene expression for the same cell while also mapping out the cell’s place in the tissue sample it came from — something that had never been done before.
This innovation had many potential applications, including enabling new ways of tracking the progression of various diseases, but there was no way to analyze all the multimodal data the method produced. In came Zhang, who became interested in designing a computational method that could.
The team focused on chromatin staining as their imaging method of choice, which is relatively cheap but still reveals a great deal of information about cells. The next step was integrating the spatial analysis techniques developed by Wang, and to do that, Zhang began designing an autoencoder.
Autoencoders are a type of neural network that typically encodes and shrinks large amounts of high-dimensional data, then expands the transformed data back to its original size. In this case, Zhang’s autoencoder did the reverse, taking the input data and making it higher-dimensional. This allowed them to combine data from different animals and remove technical variations that were not due to meaningful biological differences.
In the paper, they used this technology, abbreviated as STACI, to identify how cells and tissues reveal the progression of Alzheimer’s disease when observed under a number of spatial and imaging techniques. The model can also be used to analyze any number of diseases, Zhang says.
Xinyi with Schmidt Center fellows.
Given unlimited time and resources, her dream would be to build a fully complete model of human life. Unfortunately, both time and resources are limited. Her ambition isn’t, however, and she says she wants to keep applying her skills to solve the “most challenging questions that we don’t have the tools to answer.”
She’s currently working on wrapping up a couple of projects, one focused on studying neurodegeneration by analyzing frontal cortex imaging, and another on predicting protein images from protein sequences and chromatin imaging.
“There are still many unanswered questions,” she says. “I want to pick questions that are biologically meaningful, that help us understand things we didn’t know before.”
Adapted from an article posted on the MIT News site.
Uhler’s research lies at the intersection of machine learning, statistics, and genomics, with a particular focus on causal inference, representation learning, and gene regulation. Her use of probabilistic graphical models and development of scalable algorithms with healthcare applications has enabled her research group to gain insights into causal relationships hidden within massive amounts of data, such as those generated during gene knockout or knockdown experiments.
Caroline Uhler, director of the Schmidt Center
For almost 90 years, the title of IMS Fellow has represented a prestigious honor. Evaluated by a committee of peers, each Fellow has exhibited exceptional mastery in statistical or probabilistic research and/or has showcased remarkable leadership that has left a lasting impact on the field.
Established in 1935, the IMS is a member organization that fosters the development and dissemination of the theory and applications of statistics and probability. The IMS has over 4,700 active members throughout the world, with approximately 10% of the current IMS members earning the fellowship status. The announcement of the 2024 class of IMS Fellows can be viewed here.
In a paper published by the journal Nature Methods, Ray and Stephanie Lane Professor of Computational Biology Jian Ma and former Ph.D. students Kyle Xiong and Ruochi Zhang introduce scGHOST, a machine learning method that detects subcompartments — a specific type of 3D genome feature in the cell nucleus — and connects them to gene expression patterns. Zhang is currently a postdoctoral fellow at the Eric and Wendy Schmidt Center at the Broad Institute of MIT and Harvard.
Ruochi Zhang, postodctoral fellow at the Eric and Wendy Schmidt Center
In human cells, chromosomes aren’t arranged linearly but are folded into 3D structures. Researchers are particularly interested in 3D genome subcompartments because they reveal where chromosomes are located spatially inside the nucleus.
“One of the ultimate goals of single-cell biology is to elucidate the connections between cellular structure and function across a wide variety of biological contexts,” Ma said. “In this case, we are exploring how chromosome organization within the nucleus correlates with gene expression.”
While new technologies allow the study of these structures at the single-cell level, poor data quality can hinder precise understanding. scGHOST addresses this problem by using graph-based machine learning to enhance the data, making it easier to pinpoint and identify how chromosomes are spatially organized. scGHOST builds upon the Higashi method and its evolution, Fast Higashi, which focuses on scHi-C embeddings and imputations, that Ma's research group previously developed.
"Graph and hypergraph representation learning are integral to these methods and scGHOST, as they allow for a more nuanced and detailed exploration of the complex interactions within the genome,” said Zhang.
With the ability to accurately identify 3D genome subcompartments, scGHOST adds to the growing array of single-cell analysis tools scientists use to delineate the intricate molecular landscape of complex tissues, such as those in the brain. Ma anticipates that scGHOST could open new avenues to understanding gene regulation in health and disease.
In biomedicine, segmentation involves annotating pixels from an important structure in a medical image, like an organ or cell. Artificial intelligence models can help clinicians by highlighting pixels that may show signs of a certain disease or anomaly.
However, these models typically only provide one answer, while the problem of medical image segmentation is often far from black and white. Five expert human annotators might provide five different segmentations, perhaps disagreeing on the existence or extent of the borders of a nodule in a lung CT image.
“Having options can help in decision-making. Even just seeing that there is uncertainty in a medical image can influence someone’s decisions, so it is important to take this uncertainty into account,” says Marianne Rakic, an MIT computer science PhD candidate and fellow at the Eric and Wendy Schmidt Center at the Broad Institute of MIT and Harvard.
Rakic is lead author of a paper with others at MIT, the Broad Institute, and Massachusetts General Hospital that introduces a new AI tool that can capture the uncertainty in a medical image.
Known as Tyche (named for the Greek divinity of chance), the system provides multiple plausible segmentations that each highlight slightly different areas of a medical image. A user can specify how many options Tyche outputs and select the most appropriate one for their purpose.
Importantly, Tyche can tackle new segmentation tasks without needing to be retrained. Training is a data-intensive process that involves showing a model many examples and requires extensive machine-learning experience.
Because it doesn’t need retraining, Tyche could be easier for clinicians and biomedical researchers to use than some other methods. It could be applied “out of the box” for a variety of tasks, from identifying lesions in a lung X-ray to pinpointing anomalies in a brain MRI.
Ultimately, this system could improve diagnoses or aid in biomedical research by calling attention to potentially crucial information that other AI tools might miss.
“Ambiguity has been understudied. If your model completely misses a nodule that three experts say is there and two experts say is not, that is probably something you should pay attention to,” adds senior author Adrian Dalca, an assistant professor at Harvard Medical School and MGH, and a research scientist in the MIT Computer Science and Artificial Intelligence Laboratory (CSAIL).
Their co-authors include Hallee Wong, a graduate student in electrical engineering and computer science; Jose Javier Gonzalez Ortiz PhD ’23; Beth Cimini, associate director for bioimage analysis at the Broad Institute; and John Guttag, the Dugald C. Jackson Professor of Computer Science and Electrical Engineering. Rakic will present Tyche at the IEEE Conference on Computer Vision and Pattern Recognition, where Tyche has been selected as a highlight.
Addressing ambiguity
AI systems for medical image segmentation typically use neural networks. Loosely based on the human brain, neural networks are machine-learning models comprising many interconnected layers of nodes, or neurons, that process data.
After speaking with collaborators at the Broad Institute and MGH who use these systems, the researchers realized two major issues limit their effectiveness. The models cannot capture uncertainty and they must be retrained for even a slightly different segmentation task.
Some methods try to overcome one pitfall, but tackling both problems with a single solution has proven especially tricky, Rakic says.
“If you want to take ambiguity into account, you often have to use an extremely complicated model. With the method we propose, our goal is to make it easy to use with a relatively small model so that it can make predictions quickly,” she says.
The researchers built Tyche by modifying a straightforward neural network architecture.
A user first feeds Tyche a few examples that show the segmentation task. For instance, examples could include several images of lesions in a heart MRI that have been segmented by different human experts so the model can learn the task and see that there is ambiguity.
The researchers found that just 16 example images, called a “context set,” is enough for the model to make good predictions, but there is no limit to the number of examples one can use. The context set enables Tyche to solve new tasks without retraining.
For Tyche to capture uncertainty, the researchers modified the neural network so it outputs multiple predictions based on one medical image input and the context set. They adjusted the network’s layers so that, as data move from layer to layer, the candidate segmentations produced at each step can “talk” to each other and the examples in the context set.
In this way, the model can ensure that candidate segmentations are all a bit different, but still solve the task.
“It is like rolling dice. If your model can roll a two, three, or four, but doesn’t know you have a two and a four already, then either one might appear again,” she says.
They also modified the training process so it is rewarded by maximizing the quality of its best prediction.
If the user asked for five predictions, at the end they can see all five medical image segmentations Tyche produced, even though one might be better than the others.
The researchers also developed a version of Tyche that can be used with an existing, pretrained model for medical image segmentation. In this case, Tyche enables the model to output multiple candidates by making slight transformations to images.
Better, faster predictions
When the researchers tested Tyche with datasets of annotated medical images, they found that its predictions captured the diversity of human annotators, and that its best predictions were better than any from the baseline models. Tyche also performed faster than most models.
“Outputting multiple candidates and ensuring they are different from one another really gives you an edge,” Rakic says.
The researchers also saw that Tyche could outperform more complex models that have been trained using a large, specialized dataset.
For future work, they plan to try using a more flexible context set, perhaps including text or multiple types of images. In addition, they want to explore methods that could improve Tyche’s worst predictions and enhance the system so it can recommend the best segmentation candidates.
This research is funded, in part, by the National Institutes of Health, the Eric and Wendy Schmidt Center at the Broad Institute of MIT and Harvard, and Quanta Computer.
MURI awards support interdisciplinary teams of researchers in conducting fundamental research on topics deemed critical by the defense department. Uhler and collaborators will use the award to advance optimal intervention design in complex systems — an effort that should enhance decision-making in areas ranging from biomedical to engineering and societal applications.
Uhler, the project’s principal investigator, is a core member of the Broad Institute and a professor in the Department of Electrical Engineering and Computer Science and the Institute for Data, Systems, and Society at MIT. She is joined on the award by Alberto Abadie and Devavrat Shah, MIT; Miguel Hernan, Harvard University; John Ioannidis, Stanford University; Mengdi Wang, Princeton University; and Feng Zhang, Broad Institute. The project will involve several graduate students and postdocs.
Uhler and the research team will develop a computational framework for evidence-based decision-making. The researchers plan to host machine learning competitions to test out their methodology in engineering, biological, health, and societal application areas.
Project Title: “Evaluating, Predicting, Optimizing, and Monitoring Hypothetical Interventions in Large Networked Systems”
Neural networks have been powering breakthroughs in artificial intelligence, including the large language models that are now being used in a wide range of applications, from finance to human resources to healthcare. But these networks remain a black box whose inner workings engineers and scientists struggle to understand. Now, a team led by data and computer scientists at the University of California San Diego has given neural networks the equivalent of an X-ray to uncover how they actually learn.
The researchers found that a formula used in statistical analysis provides a streamlined mathematical description of how neural networks, such as GPT-2, a precursor to ChatGPT, learn relevant patterns in data, known as features. This formula also explains how neural networks use these relevant patterns to make predictions.
“We are trying to understand neural networks from first principles,” said Daniel Beaglehole, a PhD student in the UC San Diego Department of Computer Science and Engineering and co-first author of the study. “With our formula, one can simply interpret which features the network is using to make predictions.”
Adit Radhakrishnan, a postdoctoral fellow at Harvard who worked on the paper as an MIT EECS PhD student funded by the Schmidt Center and co-first author of the study, added: “We showed that neural networks, unlike other machine learning models, automatically implement this formula to identify features most relevant for prediction.”
The team presented their findings in the March 7 issue of the journal Science.
Why does it matter how neural networks make predictions? AI-powered tools are now pervasive in everyday life. Banks use them to approve loans. Hospitals use them to analyze medical data, such as X-rays and MRIs. Companies use them to screen job applicants. But it’s currently difficult to understand the mechanism neural networks use to make decisions and the biases in the training data that might impact this.
“If you don’t understand how neural networks learn, it’s very hard to establish whether neural networks produce reliable, accurate, and appropriate responses,” said Mikhail Belkin, the paper’s corresponding author and a professor at the UC San Diego Halicioglu Data Science Institute. “This is particularly significant given the rapid recent growth of machine learning and neural net technology.”
Former Eric and Wendy Schmidt Center PhD fellow Adit Radhakrishnan's research focuses on advancing the theoretical foundations of machine learning and developing new methods for tackling biomedical problems.
Understanding how neural networks make predictions is especially important in biological applications. In the realm of drug discovery, for example, researchers would not only want a model that accurately predicts drugs that are effective in treating cancer — they also want to discover biological mechanisms that make such drugs effective, explained Radhakrishnan. “By applying our findings to models trained to predict the effect of drugs on cancer cells, we can discover features of cancer cells that make them susceptible to a given drug and then develop new drugs to specifically target those mechanisms,” he said.
The study is part of a larger effort in Belkin’s research group to develop a mathematical theory that explains how neural networks work. “Technology has outpaced theory by a huge amount,” he said. “We need to catch up.”
The team also showed that the statistical formula they used to understand how neural networks learn, known as Average Gradient Outer Product (AGOP), could be applied to improve performance and efficiency in other types of machine learning architectures that do not include neural networks.
“If we understand the underlying mechanisms that drive neural networks, we should be able to build machine learning models that are simpler, more efficient, and more interpretable,” Belkin said. “We hope this will help democratize AI.”
The machine learning systems that Belkin envisions would need less computational power, and therefore less power from the grid, to function. These systems also would be less complex and so easier to understand.
Illustrating the new findings with an example
(Artificial) neural networks are computational tools to learn relationships between data characteristics (i.e. identifying specific objects or faces in an image). One example of a task is determining whether in a new image a person is wearing glasses or not. Machine learning approaches this problem by providing the neural network many example (training) images labeled as images of “a person wearing glasses” or ”a person not wearing glasses.” The neural network learns the relationship between images and their labels, and extracts data patterns, or features, that it needs to focus on to make a determination. One of the reasons AI systems are considered a black box is because it is often difficult to describe mathematically what criteria the systems are actually using to make their predictions, including potential biases. The new work provides a simple mathematical explanation for how the systems are learning these features.
Features are relevant patterns in the data. In the example above, there are a wide range of features that the neural networks learns, and then uses, to determine if in fact a person in a photograph is wearing glasses or not. One feature it would need to pay attention to for this task is the upper part of the face. Other features could be the eye or the nose area where glasses often rest. The network selectively pays attention to the features that it learns are relevant and then discards the other parts of the image, such as the lower part of the face, the hair and so on.
Feature learning is the ability to recognize relevant patterns in data and then use those patterns to make predictions. In the glasses example, the network learns to pay attention to the upper part of the face. In the new Science paper, the researchers identified a statistical formula that describes how the neural networks are learning features.
Alternative neural network architectures: The researchers went on to show that inserting this formula into computing systems that do not rely on neural networks allowed these systems to learn faster and more efficiently.
“How do I ignore what’s not necessary? Humans are good at this,” said Belkin. “Machines are doing the same thing. Large Language Models, for example, are implementing this ‘selective paying attention’ and we haven’t known how they do it. In our Science paper, we present a mechanism explaining at least some of how the neural nets are ‘selectively paying attention.’”
Study funders included the National Science Foundation and the Simons Foundation for the Collaboration on the Theoretical Foundations of Deep Learning. Belkin is part of NSF-funded and UC San Diego-led The Institute for Learning-enabled Optimization at Scale, or TILOS.
This interview is part of a series of short interviews from the Department of EECS, called Student Spotlights. Each Spotlight features a student answering their choice of questions about themselves and life at MIT. Today’s interviewee, Victory Yinka-Banjo, is a junior majoring in 6-7: Computer Science and Molecular Biology. Yinka-Banjo is Yinka-Banjo keeps a packed schedule; she is a member of the Office of Minority Education (OME) Laureates & Leaders program; a 2024 fellow in the public service-oriented BCAP program; has previously served as Secretary of the African Students’ Association and is now undergraduate president of the MIT Biotech Group; additionally, she is working on a cardiometabolic disease and deep learning project at the Broad Institute as an Eric and Wendy Schmidt Center Funded SuperUROP Scholar; a member of the Ginkgo Bioworks’ Cultivate Fellowship (a program that supports students interested in synthetic biology/biotech); and an ambassador for Leadership Brainery, which equips juniors/leaders of color with the resources needed to prepare for graduate school. Nevertheless, she found time to share a peek into her MIT experience with readers.
What’s your favorite building or room within MIT, and what’s special about it to you?
It has to be the Broad Institute of MIT & Harvard on Ames Street in Kendall Square, where I do my SuperUROP research in Caroline Uhler’s lab. Outside of classes, you’re 90% likely to find me on the newest mezzanine floor (between the 11th and 12th floor), in one of the UROP rooms I share with two other undergrads in the lab. We have standing desks, an amazing coffee/hot chocolate machine, external personal monitors, comfortable sofas – everything really! Not only is it my favorite building, it is also my favorite study spot on campus. In fact, I am there so often that when friends recently planned a birthday surprise for me, they told me they were considering having it at the Broad, since they could count on me being there.
I think the most beautiful thing about this building, apart from the beautiful view of Cambridge we get from being on one of the highest floors, is that when I was applying to MIT from high school, I had fantasized working at the Broad because of the ground-breaking research. To think that it is now a reality makes me appreciate every minute I spend on my floor, whether I am doing actual research or some last-minute studying for a midterm.
Tell me about one interest or hobby you’ve discovered since you came to MIT. (It doesn’t have to be academic!)
I have become pretty involved in the performing arts since I got to MIT! I have acted in two plays run by the Black Theater Guild, which was revived during my freshman year by one of my friends. I played a supporting role in the first play called Nkrumah’s Last Day, which was about Ghana at a time of governance under Kwame Nkrumah (its first president). In the second play, a ghost story/comedy called Shooting the Sheriff, I played one of the lead roles. Both caused me to step way out of my comfort zone and I loved the experiences because of that. I also got to act with some of my close friends who were first-time stage actors as well, so that made it even more fun.
Outside of acting, I also do spoken word/poetry. I have performed at events like the African Students Association Cultural Night, MIT Africa Innovate Conference and Black Womens’ Alliance Banquet. I try to use my pieces to share my experiences both within and beyond MIT, offering the perspective of an international Nigerian student. My favorite piece was called Code Switch, and I used concepts from CS & Biology (especially genetic code switching), to draw parallels with linguistic code-switching, and emphasize the beauty and originality of authenticity. This semester, I’m also a part of MIT Monologues and will be performing a piece called Inheritance, about the beauty of self-love found in affection transferred from a mother.
Are you a re-reader or a re-watcher—and if so, what are your comfort books, shows, or movies?
I don’t watch too many movies, although I used to be obsessed with all parts of High School Musical; and the only book I’ve ever reread is Americanah. I would actually say I am a re-podcaster! My go-to comfort-podcast is this episode, “A Breakthrough Unfolds”, by Google DeepMind. It makes me a little emotional every time I listen. It is such an exemplification of the power of science and its ability to break boundaries that humans formerly thought impossible. As a Computer Science & Biology major, I am particularly interested in these two disciplines’ applications to relevant problems, like the protein-folding problem discussed in the episode, which DeepMind’s solution for has caused massive advances in the biotech industry. It makes me so hopeful for the future of biology, and the ways in which computation can advance human health and precision medicine.
Who’s your favorite artist? (Using the term very broadly; any form of art can qualify!)
When I think of the word ‘artist’, I think of music artists first. There are so many who I love; my favorites also evolve over time. I’m Christian, so I listen to a lot of gospel music. I’m also Nigerian so I listen to a lot of afrobeats. Since last summer, I’ve been obsessed with Limoblaze, who fuses both gospel and afrobeats music! KB, a super talented gospel rapper, is also somewhat tied in ranking with Limo for me right now. His songs are probably ~50% of my workout playlist.
It’s time to get on the shuttle to the first Mars colony, and you can only bring one personal item. What are you going to bring along with you?
Oooh, this is a tough one, but it has to be my brass rat. Ever since I got mine at the end of sophomore year, it’s been nearly impossible for me to take it off. If there’s ever a time I forget to wear it, my finger feels off for the entire day.
Tell me about one conversation that changed the trajectory of your life.
Two specific career-defining moments come to mind. They aren’t quite conversations, but they are talks/lectures that I was deeply inspired by. The first was towards the end of high school when I watched this TEDx Talk about storing data in DNA. At the time, I was getting ready to apply to colleges and I knew that biology and computer science were two things I really liked, but I didn’t really understand the possibilities that could be birthed from them coming together as an interdisciplinary field. The TEDx talk was my eureka moment for computational biology.
The second moment was in my junior Fall during an introductory lecture to “Lab Fundamentals for Bioengineering” by Professor Jacquin Niles. I started the school year with a lot of confusion about my future post-grad, and the relevance of my planned career path to the communities that I care about. Basically, I was unsure about how Computational Biology fit into the context of Nigeria’s problems, especially because my interest in the field is oriented towards molecular biology/medicine, not necessarily public health.
In the US, most research focuses on diseases like cancer and Alzheimer’s, which, while important, are not the most pressing health conditions in tropical regions like Nigeria. When Prof Niles told us about his lab’s dedication to malaria research from a molecular biology standpoint, it was yet another eureka moment. Like yes! Computation and molecular biology can indeed mitigate diseases that affect developing nations like Nigeria–diseases that are understudied, and whose research is underfunded.
Since his talk, I found a renewed sense of purpose. Grad school isn’t the end goal. Using my skills to shine a light on the issues affecting my people that deserve far more attention is the goal. I’m so excited to see how I will use Computational Biology to possibly create the next cure to a commonly neglected tropical disease, or accelerate the diagnosis of one. Whatever it may be, I know that it will be close to home, eventually 🙂
What are you looking forward to about life after graduation? What do you think you’ll miss about MIT?
Thinking about graduating actually makes me sad. I’ve grown to love MIT. The biggest thing I’ll miss, though, is Independent Activities Period (IAP). It is such a unique part of the MIT experience. I’ve done a web development class/competition, research, a data science challenge, a molecular bio crash course, and a deep learning crash course over the past 3 IAPs. It is SUCH an amazing time to try something low stakes, forget about grades, explore Boston, build a robot, travel abroad, do less, go slower, really rejuvenate before the Spring, and embrace MIT’s motto of “mind and hand” by just being creative and explorative. It is such an exemplification of what it means to go here, and I can’t imagine it being the same anywhere else.
That said, I look forward to graduating so I can do more research. My hours spent at the Broad thinking about my UROP are always the quickest hours of my week. I love the rabbit holes my research allows me to explore, and I hope that I find those over and over again as I apply and hopefully get into PhD programs. I look forward to exploring a new city after I graduate too. I wouldn’t mind staying in Cambridge/Boston. I love it here. But I would welcome a chance to be somewhere new and embrace all the people and unique experiences it has to offer. I also hope to work on more passion projects post-grad. I feel like I have this idea in my head that once I graduate from MIT, I’ll have so much more time on my hands (we’ll see how that goes). I hope that I can use that time to work on education projects in Nigeria, which is a space I care a lot about. Generally, I want to make service more integrated in my lifestyle. I hope that post-graduation, I can prioritize doing that even more: making it a norm to lift others as I continue to climb.
Adapted from a profile posted on MIT Electrical Engineering and Computer Science Department's site.
The Eric and Wendy Schmidt Center at the Broad Institute of MIT and Harvard is delighted to announce the completion of its Cancer Immunotherapy Data Science Grand Challenge.
Participants in the challenge developed algorithms to uncover new ways to modify, or “perturb,” T cells to make them more effective at killing cancer cells. Scientists in the Hacohen Lab at the Broad then tested their predictions in mouse models, making this the first challenge that the Schmidt Center knows of in which new experiments were performed based on the output of machine-learning models developed in the challenge.
While it’s too early to say whether any of the proposed perturbations could prove useful for cancer treatment, the researchers plan to further study some of the identified perturbations and the algorithms that gave rise to them.
The Schmidt Center partnered with Harvard’s Laboratory for Innovation Science (LISH), the MIT Department of Electrical Engineering and Computer Science, Topcoder, Gordian Biotechnology, and Saturn Cloud to run the challenge. More than 1,000 people from around the world registered for the competition.
“We are thrilled that our first data science challenge attracted so many participants, including various machine-learning experts who had not previously worked on biological problems,” said Caroline Uhler, director of the Eric and Wendy Schmidt Center, a core member of the Broad Institute, and a professor in the Department of Electrical Engineering and Computer Science and the Institute for Data, Systems, and Society at MIT.
Karim Lakhani, founder and co-director of LISH and a professor of business administration at the Harvard Business School, said: “At LISH, we believe that data science challenges can help organizations harness the power of the crowd to answer pressing questions in biology and other fields. We hope this challenge will serve as a case study in how machine-learning experts can collaborate with biologists to improve experimental design.”
Boosting cancer research with machine learning
Cancer immunotherapy seeks to harness the body’s immune system, and most often T cells, to recognize and kill cancer cells while leaving healthy cells alone. In the last decade, there have been many breakthroughs in cancer immunotherapy, yet treatments still only work for some cancer patients some of the time.
“We’re hopeful that challenges like this can help us home in on T-cell-perturbations that could ultimately lead to new therapeutics — and make cancer immunotherapy work for more patients,” said Nir Hacohen, an institute member at Broad, director of the Broad Institute’s Cell Circuits Program, and director of the Center for Cancer Immunology at Mass General Brigham.
Marc Schwartz, a postdoctoral fellow in the Hacohen Lab, previously ran experiments testing the effects of 73 gene knockouts in T cells in mouse models. Because researchers can’t scale mouse model experiments beyond 100 or so genes at a time, it’s not feasible to test out every gene in a particular disease pathway, explained Schwartz.
“That’s why we were excited about the idea of testing a limited number of genes that we think are important and then training an algorithm to learn something that we can't see from that data on our own,” he added.
The overarching data science challenge was divided into three parts that ran as individual data science competitions on Topcoder. In Challenge 1, participants received gene expression data from 66 of the 73 T-cell-gene knockouts from Schwartz’s experiments as training data. They then developed an algorithm that could predict how knocking out the seven “held-out” genes would affect T cells.
Challenge 2 participants used their algorithms from the first challenge to propose new gene knockouts (picking from any of the 20,000 genes in the entire genome) to shift as many T cells as possible into a cancer-fighting state. In Challenge 3, participants proposed a metric for ranking how well a particular gene knockout would bring about this desired shift in T cells.
To make the challenge accessible to participants without a biology background, Orr Ashenberg, associate director of computational biology at the Klarman Cell Observatory of the Broad Institute, produced a 1.5-hour crash course on cancer biology, genetic perturbations, and single-cell sequencing technologies.
Orr Ashenberg, associate director of computational biology at the Broad's Klarman Cell Observatory, delivers a lecture on single-cell sequencing technologies.
The Schmidt Center announced the Challenges 1 and 3 winners last March. The researchers then ran the top-scoring algorithms from Challenge 1 to predict which genes to knock out to mimic two kinds of cancer immunotherapy — CAR T-cell therapy and checkpoint blockade therapy. Next, Schwartz conducted experiments to see how well the proposed gene knockouts performed in a mouse model. To determine the Challenge 2 winners, Schmidt Center research fellow Jiaqi Zhang, who was instrumental in developing the challenge, calculated how well each participant’s algorithm from Challenge 1 predicted the effects of those ~60 gene knockouts.
The winners of Challenge 2 — the final part of the competition — are:
-First place: Brody Langille, Jordan Trajkovski, and Elizabeth Hudson
-Second place: mglettig (username)*
-Third place: Ai Vu Hong, researcher at Genethon, France
-Sixth place: John Gardner, freelance data scientist
-Seventh place: agilsoft (username)*
-Eighth place: Basak Eraslan, postdoctoral researcher holding a joint position at the Regev Lab in Genentech and Kundaje Lab at Stanford University
-Ninth place: Haoyue Dai, Kun Zhang, Ignavier Ng, Yujia Zheng, Xinshuai Dong, and Yewen Fan from Carnegie Mellon University; Petar Stojanov, postdoctoral fellow at the Eric and Wendy Schmidt Center; Gongxu Luo, Mohamed bin Zayed University of Artificial Intelligence; and Biwei Huang, University of California, San Diego
-Ninth place: Liu Xindi, freelance programmer
-Ninth place: Johnson Zhou, Camille Sayoc, and Yi-Cheng Peng, Master’s students of the Faculty of Engineering and IT at the University of Melbourne, Victoria, Australia
The winning teams approached the problem using different deep-learning methods depending on the chosen input features. These features include gene expression and “chromatin accessibility,” the degree to which genetic information encoded in DNA can be accessed and read, measured by ATAC-seq peak counts. Additionally, some of the top-scoring teams incorporated learned representations from variational autoencoders — models that can capture meaningful features from raw data — or graph neural networks constructed based on the gene ontology database.
"We are grateful for the opportunity to participate in this challenge and are excited by the results,” said the first-place team in a prepared statement. “It's not often that you get invited to work on an important problem alongside preeminent scientists who furnish the problem description and data that you need to develop a novel solution — a novel solution that those same scientists can then turn around and validate in their lab.”
Martin Borch Jensen, chief scientific officer of Gordian Biotechnology, said: "Technological advances in sequencing have led to a vast amount of genomics data. As we pile up more and more transcriptomes from every type of cell in the human body, it becomes increasingly valuable to develop ways to understand how gene expression can cause and predict health and disease. I'm very excited for this competition to catalyze more work on this problem.”
Now, researchers at the Schmidt Center will further study the top-scoring algorithms to see if they can combine components from each into an even better predictive tool. The center plans to hold its second data science challenge later this year.
*Editor's note: Usernames were used instead of participant names in cases where the Schmidt Center could not get in touch with winners.
In a Comment for Nature Cell Biology, the Eric and Wendy Schmidt Center's director Caroline Uhlerdiscusses how the rise of large-scale datasets in biology positions the field to become a driver of foundational advances in machine learning — and vice versa. Uhler, who is also a full professor in the Department of Electrical Engineering and Computer Science and the Institute for Data, Systems, and Society at MIT, advocates for new machine learning models that can better integrate different types of biological data and can uncover causal mechanisms in disease, not just associations. She also discusses the need for close collaborations between biologists and computational scientists so that predictive and causal algorithms can be incorporated into experimental design — and outlines some of the challenges, such as distinct cultures and vocabularies, of building those teams.
As we age, the risk for a wide range of diseases, including cancer and neurodegenerative conditions, increases. But while aging has been extensively studied, scientists don’t have a clear picture of the molecular changes that take place as we get older.
Now, researchers at the Broad Institute of MIT and Harvard and ETH Zürich in Switzerland have found key gene-expression regulators related to cellular aging that are tightly coupled to structural alterations of chromatin — the DNA-protein complex that forms chromosomes. The findings, published last month in Aging Cell, offer new insights into the biology of cellular aging. The research may also provide potential targets for aging reversal.
The study stems from a long-term collaboration between the laboratory of GV Shivashankar at ETH Zürich on the biological side and Caroline Uhler at the Broad Institute on the computational side.
“The explosion of biomedical data presents an exciting opportunity to develop novel machine learning methods to help answer important biological questions,” said study co-senior author Uhler, the director of the Eric and Wendy Schmidt Center at the Broad and a professor in the Department of Electrical Engineering and Computer Science and the Institute for Data, Systems, and Society at MIT. “In this work, the availability of large-scale sequencing data from many individuals in different age groups motivated us to develop methods to identify drivers of cellular aging,” she added.
Shivashankar’s lab has long been interested in understanding the relationship between a cell’s microenvironment, the three-dimensional structure of the genome, and gene expression in health and disease. Depending on how DNA is packed inside a cell’s nucleus, it may alter the expression of specific genes, which could in turn result in certain diseases, explained co-senior author Shivashankar, professor of Mechano-Genomics at ETH Zürich and head of the Laboratory of Nanoscale Biology at the Paul Scherrer Institute in Switzerland. “We’re very excited about understanding what may lead to healthy aging as opposed to cancer or neurodegeneration,” he added.
The study also aligns with the Eric and Wendy Schmidt Center’s goal of developing computational approaches for challenging biomedical questions. To this end, the Schmidt Center trains talented undergraduate, master’s, and PhD students as well as postdoctoral fellows with computational backgrounds on how to work with experimental biologists.
“As a graduate student in statistics, working closely with a biological lab allows me to gain a much deeper understanding of the kinds of questions and data that are most interesting to biologists,” said study co-first author Louis Cammarata, a PhD student at Harvard University and the Eric and Wendy Schmidt Center. “I’m able to design more useful computational methods because of this constant communication.”
Drivers of aging
In the nucleus of a cell, DNA coils around proteins to form chromatin. Other proteins bind along chromatin, creating complex three-dimensional structures that leave some genes accessible to transcription and others closed off.
Clockwise from top right: Caroline Uhler, GV Shivashankar (image credit: Paul Scherrer Institute), Louis Cammarata, and Jana Braunger
Uhler, Shivashankar, and their teams analyzed gene expression data from skin cells of 133 individuals aged 1 to 96 years, who were divided in five age groups. The difference in gene expression was particularly prominent when comparing the two oldest groups, which included people aged 61 to 85 years and those aged 86 to 96 years. Differentially expressed genes tended to be involved in biological processes such as immune response and cell proliferation, which play important roles in aging.
Next, the researchers used statistical algorithms to combine these data with information from a database that lists protein-protein interactions. The analysis revealed key age-associated regulators of gene expression, which include transcription factors — proteins that control how other genes are expressed.
“Transcription factors may be post-translationally activated or they may benefit from changes in chromatin organization to activate their target genes at a later time point,” said study co-first author Jana Braunger, a former master’s student at the Eric and Wendy Schmidt Center and current PhD student at the University of Heidelberg.
Gene expression hubs
To analyze the coupling between chromatin organization and changes in gene expression, the researchers used an experimental method called Hi-C, which provides a proximity map of the DNA packing.
Comparing Hi-C data from old and young skin cells revealed that the structure of chromatin changes over time, either drawing apart genes that were close together or bringing together genes that were far apart in young cells.
In the cell’s nucleus, nearby genes are often expressed as a group, Cammarata explained. “There are specific hotspots where different chromosomes come together, along with other molecules that are useful for transcription, and within those hubs, you have active transcription and co-regulation of genes,” he said. “In aging, changes in how DNA is folded influence these hotspots of transcription.”
Mitigating aging
Although more work is needed to determine whether alterations in chromatin structure drive changes in gene expression or vice versa, some of the gene-expression regulators identified in this study could serve as potential targets to mitigate, prevent, or even reverse cellular aging. “Identifying the key transcriptional drivers of cellular aging is crucial to develop interventions for cellular reprogramming and rejuvenation,” Shivashankar said.
Uhler noted that the study is an example of how computational researchers can develop new methods to help answer important biological questions — a core mission of the Eric and Wendy Schmidt Center. “We place great importance on training the next generation of scientists — researchers who are strong on the computational side and understand the biological questions,”she said. “Merging computational science and biology can help us tackle some of medicine’s biggest challenges.”
In 2003, scientists finished sequencing almost all of the three million nucleotide base pairs that make up the human genome. This feat led to an explosion in genomics analysis, which to this day relies on aligning sequencing data to a “reference genome” — a composite made up of DNA samples from different individuals in the same species — for humans and other species.
Now, researchers at Stanford University and the Eric and Wendy Schmidt Center at the Broad Institute of MIT and Harvard have developed a genomics analysis framework, SPLASH, that directly analyzes raw sequencing samples, eliminating the need for reference data. The method can perform genomic analyses more quickly and with less computing power than traditional methods. SPLASH should prove especially useful for analyzing genomes of understudied or rapidly mutating species.
In a study published earlier this month in Cell, the team showed that the framework can detect different strains of SARS-CoV-2 and find sequence diversity in adaptive immune receptors, among other findings. Kaitlin Chuang and Tavor Baharav, former PhD students at Stanford University, were co-first authors on the paper, and Julia Salzman, associate professor of biomedical science and biochemistry at Stanford, was the lead author. All research was performed in Salzman's group, whose lab combines statistics and genomics.
“A lot of sequencing analysis is done with implicit priors, meaning that your pipeline is only going to identify the one feature that it was designed to find,” said Baharav, who is now an Eric and Wendy Schmidt Center postdoctoral fellow. “With SPLASH, we’ve developed a method for unbiased, reference-free hypothesis generation.”
From alignment- to statistics-first
While genomics has revolutionized both medicine and ecology, its dependence on reference genomes has its limitations. For example, only 5% of mammalian species have had their genomes sequenced — a percentage that drops even further for organisms like bacteria and viruses. Additionally, because the human-reference genome only contains samples from a handful of individuals, it does not reflect global genomic diversity.
Eric and Wendy Schmidt Center postdoctoral fellow Tavor Baharav
Also, traditional genomics analysis aligns samples with references before comparing the samples to each other, discarding outliers. “When you're trying to detect an interesting, novel event, it almost by definition isn't going to align well to the reference,” said Baharav.
To address these and other limitations, researchers in the Salzman Lab at Stanford University came up with a way to analyze raw sequencing data without having to first align it to a reference genome.
Their framework, SPLASH, identifies unchanging "anchor" subsequences in the raw sequencing data that are followed by "target" sequences that vary by sample. SPLASH, which stands for “Statistically Primary aLignment Agnostic Sequence Homing,” uses a new statistical test to determine which stretch of RNA reads exhibit the most variation.
"This work illustrates how interdisciplinary teams with diverse perspectives and skill sets are powerful and needed for scientific progress,” said Salzman. “Initially, the team questioned why such a straightforward approach hadn't been implemented before, but we gradually came to realize that rethinking conventions can sometimes yield simple solutions that could work better than ingrained approaches.”
Unlike traditional methods, which can only detect certain types of genetic variations, the framework can detect a wide variety of variations. SPLASH is also much more computationally efficient than those methods. An updated version of the framework can complete the entire analysis in an hour while using much less computing power than alignment-first approaches.
Detecting viral mutations + microalgae growing on eelgrass
To test the effectiveness of SPLASH, the team used it to perform a range of genomic analyses. In one, they compared nasal swab samples from patients taken at different periods during the COVID-19 pandemic, when different viral strains were dominant. SPLASH was able to identify which anchors had “low p-values” and high effect sizes — indicators of viral mutations. They then mapped these reads to control samples from different COVID strains, determining that almost all of the anchors that SPLASH homed in on were indeed strain-defining mutations.
Eelgrass provides foraging areas and shelter for fish. Adam Obaza/NOAA.
Given that very few species have reference genomes, the team also tested how well SPLASH can detect variations between samples from two species — eelgrass and octopus — with limited reference data available. They compared RNA from eelgrass, a common seagrass, found in the Mediterranean and Norway, finding that almost 6% of targets did not align to eelgrass references. In particular, they noticed that the target sequences for one anchor varied by location and season.
The team theorized that these discrepancies could indicate the presence of different species of diatoms, microalgae that grow on other plants, as the anchor was less abundant in samples taken at night, when diatoms reduce expression of this particular type of gene.
“On its own, SPLASH does not provide immediately interpretable results, but it points researchers to interesting questions that they can investigate further,” said Baharav.
Next steps
Baharav, who completed his PhD in electrical engineering at Stanford earlier this year, is now applying his computational background to cancer research. As white blood cells develop, they shuffle around parts of their genome through a process called “V(D)J recombination.” This genetic reshuffling allows them to produce a huge array of antibodies and T-cell receptors, which they use to recognize and kill millions of microbes.
Cancer researchers like Baharav’s mentor, Rafael Irizarry, chair of the Department of Data Science at Dana-Farber Cancer Institute, want to better understand how V(D)J recombination works to design cancer vaccines. As a Schmidt Center fellow, Baharav is developing a reference-free way to analyze these adaptive immune receptors.
“SPLASH provides an exciting new statistical and computational framework for genomic analysis. I'm looking forward to building on this work to expand the scope of reference-free analysis, allowing researchers to perform unbiased inference on their data,” said Baharav. “As discussed in SPLASH, reference-based methods fall short in analyzing highly diverse genomic regions such as T cell receptors, which I'm looking to change.”
The Eric and Wendy Schmidt Center at the Broad Institute of MIT and Harvard is excited to announce that postdoctoral fellow Maria Skoularidou was awarded the 2023 Blackwell-Rosenbluth Award earlier this month. The Blackwell-Rosenbluth Award is granted to outstanding young researchers in the field of Bayesian statistics.
Skoularidou joined the Eric and Wendy Schmidt Center in September, 2023. She is co-advised by Nikos Daskalakis, director of the Neurogenomics and Translational Bioinformatics Laboratory at McLean Hospital and an associate professor of psychiatry at Harvard Medical School, and Costis Daskalakis, a professor in MIT’s Department of Electrical Engineering and Computer Science and a member of the MIT Computer Science and Artificial Intelligence Laboratory. Her research focuses on developing scalable and efficient computational methods to detect epigenetic effects in diverse trauma and PTSD contextsthrough employing information from various datasets.
Skoularidou holds a PhD in biostatistics from the University of Cambridge, where she was advised by Sylvia Richardson. Skoularidou has a four-year degree in informatics and a Master’s of Science in statistical science from the Athens University of Economics and Business. She founded (Dis)Ability in AI, a group that supports and advocates for disabled people’s needs at machine learning conferences and other venues, and is on the editorial board of ACM Transactions on Probabilistic Machine Learning.
“Maria has already made impressive contributions to the field of Bayesian inference as well as generative modeling and its applications to biomedical data,” said Caroline Uhler, director of the Eric and Wendy Schmidt Center, a core member of the Broad Institute, and a professor in the Department of Electrical Engineering and Computer Science and the Institute for Data, Systems and Society at MIT. “We’re excited to see what she’ll continue to accomplish as a Schmidt Center fellow.”
A strategy for cellular reprogramming involves using targeted genetic interventions to engineer a cell into a new state. The technique holds great promise in immunotherapy, for instance, where researchers could reprogram a patient’s T-cells so they are more potent cancer killers. Someday, the approach could also help identify life-saving cancer treatments or regenerative therapies that repair disease-ravaged organs.
But the human body has about 20,000 genes, and a genetic perturbation could be on a combination of genes or on any of the over 1,000 transcription factors that regulate the genes. Because the search space is vast and genetic experiments are costly, scientists often struggle to find the ideal perturbation for their particular application.
Researchers from MIT and Harvard University developed a new, computational approach that can efficiently identify optimal genetic perturbations based on a much smaller number of experiments than traditional methods.
Their algorithmic technique leverages the cause-and-effect relationship between factors in a complex system, such as genome regulation, to prioritize the best intervention in each round of sequential experiments.
The researchers conducted a rigorous theoretical analysis to determine that their technique did, indeed, identify optimal interventions. With that theoretical framework in place, they applied the algorithms to real biological data designed to mimic a cellular reprogramming experiment. Their algorithms were the most efficient and effective.
“Too often, large-scale experiments are designed empirically. A careful causal framework for sequential experimentation may allow identifying optimal interventions with fewer trials, thereby reducing experimental costs,” says co-senior author Caroline Uhler, a professor in the Department of Electrical Engineering and Computer Science (EECS) who is also the director of the Eric and Wendy Schmidt Center at the Broad Institute of MIT and Harvard, and a researcher at MIT’s Laboratory for Information and Decision Systems (LIDS) and Institute for Data, Systems and Society (IDSS).
Joining Uhler on the paper, which appears today in Nature Machine Intelligence, are lead author Jiaqi Zhang, a graduate student and Eric and Wendy Schmidt Center Fellow; co-senior author Themistoklis P. Sapsis, professor of mechanical and ocean engineering at MIT and a member of IDSS; and others at Harvard and MIT.
Active learning
When scientists try to design an effective intervention for a complex system, like in cellular reprogramming, they often perform experiments sequentially. Such settings are ideally suited for the use of a machine-learning approach called active learning. Data samples are collected and used to learn a model of the system that incorporates the knowledge gathered so far. From this model, an acquisition function is designed — an equation that evaluates all potential interventions and picks the best one to test in the next trial.
This process is repeated until an optimal intervention is identified (or resources to fund subsequent experiments run out).
“While there are several generic acquisition functions to sequentially design experiments, these are not effective for problems of such complexity, leading to very slow convergence,” Sapsis explains.
Acquisition functions typically consider correlation between factors, such as which genes are co-expressed. But focusing only on correlation ignores the regulatory relationships or causal structure of the system. For instance, a genetic intervention can only affect the expression of downstream genes, but a correlation-based approach would not be able to distinguish between genes that are upstream or downstream.
“You can learn some of this causal knowledge from the data and use that to design an intervention more efficiently,” Zhang explains.
The MIT and Harvard researchers leveraged this underlying causal structure for their technique. First, they carefully constructed an algorithm so it can only learn models of the system that account for causal relationships.
Then the researchers designed the acquisition function so it automatically evaluates interventions using information on these causal relationships. They crafted this function so it prioritizes the most informative interventions, meaning those most likely to lead to the optimal intervention in subsequent experiments.
“By considering causal models instead of correlation-based models, we can already rule out certain interventions. Then, whenever you get new data, you can learn a more accurate causal model and thereby further shrink the space of interventions,” Uhler explains.
This smaller search space, coupled with the acquisition function’s special focus on the most informative interventions, is what makes their approach so efficient.
The researchers further improved their acquisition function using a technique known as output weighting, inspired by the study of extreme events in complex systems. This method carefully emphasizes interventions that are likely to be closer to the optimal intervention.
“Essentially, we view an optimal intervention as an ‘extreme event’ within the space of all possible, suboptimal interventions and use some of the ideas we have developed for these problems,” Sapsis says.
Enhanced efficiency
They tested their algorithms using real biological data in a simulated cellular reprogramming experiment. For this test, they sought a genetic perturbation that would result in a desired shift in average gene expression. Their acquisition functions consistently identified better interventions than baseline methods through every step in the multi-stage experiment.
“If you cut the experiment off at any stage, ours would still be more efficient than the baselines. This means you could run fewer experiments and get the same or better results,” Zhang says.
The researchers are currently working with experimentalists to apply their technique toward cellular reprogramming in the lab.
Their approach could also be applied to problems outside genomics, such as identifying optimal prices for consumer products or enabling optimal feedback control in fluid mechanics applications.
In the future, they plan to enhance their technique for optimizations beyond those that seek to match a desired mean. In addition, their method assumes that scientists already understand the causal relationships in their system, but future work could explore how to use AI to learn that information, as well.
This work was funded, in part, by the Office of Naval Research, the MIT-IBM Watson AI Lab, the MIT J-Clinic for Machine Learning and Health, the Eric and Wendy Schmidt Center at the Broad Institute, a Simons Investigator Award, the Air Force Office of Scientific Research, and a National Science Foundation Graduate Fellowship.
Adapted from a news story posted on the MIT News website.
Scientists using machine learning tools to analyze biomedical data often turn to neural network algorithms, but before these models became popular, another simpler type of machine learning algorithm called kernel methods were commonly used. Kernel methods work by first applying straightforward operations to transform data and then training a simple model on the transformed data.
Now, in a new paper recently published in Nature Communications, researchers at the Eric and Wendy Schmidt Center at the Broad Institute of MIT and Harvard have developed a new way of using kernel methods that could make them more useful for a wider range of applications, such as virtual drug screening. They came up with the first “transfer learning” techniques for kernel methods that can be successfully applied to large-scale datasets. Transfer learning allows researchers to improve machine learning models by training them on one task in a way that enhances their performance on a second task — without having to spend the time and resources training a new model for each new task. In their paper, the team showed how their transfer learning framework allowed them to predict which drugs might be most effective in certain cancer cell lines where little data is available. They did this by transferring from cell lines in which many drugs have already been tested.
“Before our paper, there was no transfer learning method for kernel methods that could scale to the large datasets of most interest in the biomedical field and beyond. We’ve shown for the first time that transfer learning using kernels in these settings is possible and I think that is really exciting,” said Caroline Uhler, the senior author on the paper and a Broad core institute member, co-director of the Schmidt Center at Broad, and a professor in the Department of Electrical Engineering and Computer Science as well as the Institute for Data, Systems, and Society at MIT.
The team’s key innovation was creatively adapting transfer learning methods used in neural network algorithms so that they can be applied to kernel methods. This advance could find uses in other applications.
“Particularly for healthcare and biomedical applications, it's very hard to collect a lot of data for every question of interest. When you have very little data for a certain task but a related task has abundant data, this is exactly a setting where our method is effective,” said Adityanarayanan Radhakrishnan, a co-first author on the study and a Schmidt Center fellow, who worked on this study while completing his PhD as an Eric and Wendy Schmidt Center Fellow in Uhler’s lab at Broad and MIT, and is currently the George F. Carrier Postdoctoral Fellow at Harvard School of Engineering and Applied Sciences.
Transferring knowledge
The research team focused on kernel methods because they found in a previous paper that these performed better than typical neural network models on virtual drug screening tasks. But they wanted to make it possible for researchers to quickly reuse their kernel method algorithms to identify drugs for a wide range of cancer types without having to train a new model for each new type of cancer. They realized that transfer learning techniques are necessary for this, but because existing techniques don’t work well for kernel methods, they had to come up with new ones.
They decided to take inspiration from two transfer learning techniques that work well for neural network models, which they called projection and translation. The team adapted them to work with kernel methods and then tested their approach in a virtual drug screen.
The researchers analyzed performance of their transfer learning algorithms on two massive Broad datasets, one from the Connectivity Map (CMAP) and the other from the Cancer Dependency Map (DepMap). These datasets describe the effects of drugs on cancer cell lines across millions of drug and cell line combinations. The team trained their kernel method algorithms to predict either the genes expressed by a certain cell type after it was treated with a certain drug (using the CMAP dataset), or the proportion of cancer cells that survived after treatment with the same drug (using the DepMap dataset).
The scientists then applied their projection and translation techniques to their model so that it could complete the second task: to predict the effect of the drug on new cancer cell lines that have much less data. The projection transformation corrects the model’s predictions on the second task by recognizing when the prediction errors are falling into categories that can be easily corrected to the right category. And the translation technique fine-tunes the model by applying a correction term that shifts the model’s predictions so that it’s more accurate on the second task.
The team found that their transfer learning techniques allowed their original kernel method to be successfully “transferred” to the second task, without needing to be retrained. Compared to a new model trained only on the second task, the transfer learning techniques greatly boosted the accuracy of their model in predicting the effect of drugs for new cancer cell lines. And on a common machine learning task where the team trained their kernel method algorithms to recognize images, their approach surprisingly boosted the accuracy by up to 10 percent.
Moreover, the researchers were also able to pinpoint exactly how much extra data they would need to collect to increase the performance of the model. Uhler said this could be helpful to scientists trying to decide whether it’s worthwhile to collect more data in the lab. “That's really quite exciting because you can ask ‘how much is it worth for me to have a little bit better performance of my model if I know that we’ll need to collect, say, 10 or 20 percent more data?’” said Uhler.
Beyond drug screening
Two additional advantages of kernel methods are that they provide interpretability as well as a quantification of how uncertain the model is on a given prediction. To take advantage of the interpretability aspect, the research team is working on pinning down the features of a drug that lead their model to predict that it will be effective. In addition, the research team hopes that the uncertainty estimates provided by their kernel approach will be helpful in identifying which new drug and cell line combinations should be screened experimentally for a more effective drug discovery pipeline.
They also have plans to expand their framework to other applications, such as screening cancer genes that tumors heavily depend on for survival and might be targeted with new drugs.
The team adds that their transfer learning approach for kernel methods may also open up other, unexpected applications. Because kernel methods make it easy for scientists to mathematically understand what the model is doing, they can investigate what kinds of biomedical questions will be the best fit to study. “It now gives us a more thorough or deeper understanding of transfer learning and where the power comes from, so that we can analyze which tasks it will actually work for,” said Uhler.
Helmholtz Munich and the Eric and Wendy Schmidt Center at the Broad Institute of MIT and Harvard today announce the launch of a collaboration to bridge a gap in health research with AI and machine learning.
In the past decade, the field of genomics has accelerated to a point where we can now both measure and perturb biological systems at massive, unprecedented scales, holding huge potential for disease treatment. However, the computational tools needed to take advantage of all this data have not kept pace. By leveraging machine learning methods, the partnership between Helmholtz Munich and the Eric and Wendy Schmidt Center seeks to gain valuable insights into important genomics problems while simultaneously advancing the foundations of machine learning through novel research inspired by genomics questions.
Leading this joint initiative are Caroline Uhler, co-director of the Eric and Wendy Schmidt Center at the Broad Institute, and Fabian Theis, head of the Computational Health Center (CHC) at Helmholtz Munich and Director of Helmholtz AI. Both Caroline Uhler and Fabian Theis have backgrounds in machine learning, statistics, data science, biology, and human biology. “This exchange model between the Broad Institute and Helmholtz Munich will merge our expertise on machine learning and genomics to foster innovative ways to address major challenges in biomedical research,” said Fabian Theis.
The collaboration will encompass a range of activities, including the exchange of graduate students, postdoctoral fellows, and other research staff between the two research centers. These individuals will undertake short research stays, enabling them to benefit from the expertise and resources available at both centers. In addition, the research centers will co-organize workshops and conferences to facilitate knowledge exchange and foster collaboration in the field of AI and genomics.
“Despite an explosion in biological data, the technology sector remains the key driver of machine learning advances today,” said Caroline Uhler. “Both Helmholtz Munich and the Broad Institute are seeking to change that by developing foundations of machine learning that are geared specifically to biological problems, and we’re excited for this collaboration to amplify our efforts.”
Gemma Moran will never forget how magical it felt to run her very first statistical models on genomics data during her undergraduate summer research project at the University of Sydney. Moran had initially planned to major in pure mathematics but veered away from that path towards a career in applied research after taking a few statistics courses. “I came to realize that I was much more interested in being able to apply math to real world applications and data,” she said.
Now, as a postdoctoral fellow with the Eric and Wendy Schmidt Center at the Broad Institute of MIT and Harvard, Moran’s interest in using statistical models to uncover biological patterns that could improve health care has only grown stronger. These days, Moran, who is based at Columbia University’s Data Science Institute, is working to combine the rigorous and intuitive nature of the simple statistical models she first learned about in undergrad with the flexibility and power of today’s modern machine learning algorithms. In September, Moran will launch her own research group to pursue this direction as an assistant professor of statistics at Rutgers University.
Those who work with her are confident that her research has been and will continue to be impactful. “Gemma is a clear thinker, a careful scientist, and a fantastic collaborator to work with and learn from,” said David Blei, Moran’s postdoctoral adviser and a professor of statistics and computer science at Columbia University. “What her algorithms discover is information that we can use to help make better scientific and medical predictions, and use to help further our understanding of biology and genetics.”
As part of a project with Anthony Philippakis, co-director of the Eric and Wendy Schmidt Center and chief data officer of the Broad Institute, Moran has been using a type of machine learning algorithm called a variational autoencoder (VAE) to reveal important connections between disease symptoms that doctors may be missing. Though it’s still in its early stages, this work has the potential to affect clinical care if these algorithms discover new ways to cluster symptoms into one disease versus another — a challenging task that has long relied on doctor’s observations alone.
Revealing New Relationships
As a graduate student at the University of Pennsylvania, Moran worked on designing a method to uncover genes that are most relevant in different subtypes of breast cancer. She also developed new theoretical techniques to estimate the uncertainty present in models. During her postdoctoral fellowship in Blei’s lab, Moran developed a new method that allows researchers to better interpret the results that variational autoencoder algorithms spit out. These algorithms are masterful at paring down massive datasets into tiny summaries that contain only the most important aspects of the bigger dataset. The problem, Moran explains, is that it’s very challenging for researchers to understand exactly what parts of the original dataset are captured in the small summaries.
Moran working in her office in Columbia's Data Science Institute
To illustrate the challenge and her new fix, Moran gives the example of a large dataset filled with hundreds and hundreds of movie ratings. To create a meaningful summary with fewer data points, the variational autoencoder algorithm might divide these ratings into categories like horror, comedy, action, and science fiction. While it learns, the algorithm creates connections between the movie titles in the original dataset and its new summary output. But if left to its own devices, the algorithm will create thousands of connections that will be difficult to interpret.
Importantly, by pruning down these connections at certain places in the network until they become sparse, Moran’s new method — named "sparse VAE" — makes it much easier to see what parts of the original data are directly linked to the smaller summary. For example, she could trace back the new “anchor points” to find that the movie “Alien” is only represented in the science fiction category of the summary, but a movie like “Everything Everywhere All At Once” might be represented in the categories of action, comedy, and science fiction. And as an added rare bonus, Moran’s new method successfully achieves a statistical property known as identifiability. This ensures that the model only has one way to interpret it, as long as there are anchor points in the data.
After chatting with Philippakis last year about her new sparse VAE method, the two realized that it could be a great way to unearth previously unknown relationships between health symptoms in ways that would be easy for doctors and health researchers to interpret. Essentially, their project uses machine learning to improve nosology, which is the scientific field of disease classification. Until now, to classify a new disease, doctors have relied on their own expertise and experience to know what symptoms — like blurry vision and increased urination for diabetes — co-occur. They’ve also had to decide how to meaningfully differentiate these symptoms from another group of symptoms that comprise a separate disease. But it’s possible that physicians haven’t noticed some co-occurring symptoms that might tell them more about disease severity or indicate a new subgroup of a disease — or require a new disease label altogether.
“What these machine learning methods are exactly designed to do is find what things travel together, and so in that way, they can help physicians see more things that travel together that they might not have noticed just by observation alone,” said Moran.
Moran stands in front of the Low Memorial Library
Moran and Philippakis are currently applying the sparse VAE method to data from 500,000 patients in the UK Biobank, which is a large patient dataset filled with detailed genetic and health information collected by researchers in the United Kingdom. They hope it may yield surprising correlations between biological signals that could improve the classification of diseases, with the goal of obtaining their first results later this year.
“I’m incredibly excited about where this line of research is headed,” said Anthony Philippakis. “In the same way that Gemma has already shown that her method can identify ‘eigen-movies’ that indicate similar classes of films, there is the opportunity to uncover ‘eigen-phenotypes’ that indicate collections of traits that are correlated with each other.”
New Job, Same Thrill
When Moran starts her own research group this fall at Rutgers University, she will continue her work on improving the interpretability and transparency of powerful machine learning algorithms applied to medical research. Her ultimate goal is to create algorithms that provide the most advantages to the health of society without propagating harmful biases against certain groups. Indeed, Moran sees this problem of bias in machine learning as one of the biggest challenges facing the field over the next ten years.
“It’s a really crazy time to be in machine learning. There are so many developments happening at breakneck speed,” she said. “What worries me is people building these powerful [machine learning] models without necessary checks and balances and transparency and interpretability … especially applied to health care because it's such a critical domain where we could see negative consequences if we're not using these tools responsibly.”
While Moran’s goals and physical locations on opposite sides of the globe have changed across her academic career, the joy she finds in the work has remained constant. “That feeling when you've had an idea and then you code up something that works — it's just very thrilling,” she said. For Moran, that thrill becomes even more meaningful when she’s answering a question that could help actual patients. “At the end of the day, I love math and modeling and thinking about variation and how to think about data, but it's nice to connect it to real world questions.”
Eric and Wendy Schmidt Center at the Broad Institute of MIT and Harvard is excited to announce that postdoctoral fellow Yue Qin was named to the Forbes 30 Under 30 Asia 2023 list this May. The Forbes 30 under 30 lists highlight some of the most successful researchers, leaders, and entrepreneurs around the world.
Qin joined the Eric and Wendy Schmidt Center in January, 2023. She is co-advised by Paul Blainey, a core member of the Broad Institute and an associate professor of biological engineering at MIT, and Caroline Uhler, co-director of the Eric and Wendy Schmidt Center. Qin's research interests lie in understanding how to read out the programs of cells from the genome. Qin uses that knowledge to create in silico cells that simulate the effect of therapeutic interventions in different disease and genetic contexts with the ultimate goal of developing personalized medicine.
Qin holds a PhD in Bioinformatics and Systems Biology and a BSc in Bioinformatics from the University of California San Diego (UCSD). As a graduate student, she was the first author on a 2021 Nature paper that developed a machine learning framework to map the structure of human cells by fusing data from protein imaging and protein biophysical interactions. Qin is a Siebel Scholar and a recipient of an NCI Predoctoral to Postdoctoral Fellow Transition Award (F99/K00) as well as the Chancellor’s Dissertation Medal within the Jacobs School of Engineering at UCSD.
“Yue embodies the type of researcher we’re excited to work with at the Eric and Wendy Schmidt Center,” said Uhler, who is also a core member of the Broad Institute and a professor in the Department of Electrical Engineering and Computer Science and the Institute for Data, Systems and Society at MIT. “Her research is a great example of how computation and biology can go hand in hand in an age where the number of possible experiments we could perform has exploded.”
To get an inside look at the heart, cardiologists often use electrocardiograms (ECGs) to trace its electrical activity and magnetic resonance images (MRIs) to map its structure. Because the two types of data reveal different details about the heart, physicians typically study them separately to diagnose heart conditions.
Now, in a paper published in Nature Communications, scientists in the Eric and Wendy Schmidt Center at the Broad Institute of MIT and Harvard have developed a machine learning approach that can learn patterns from ECGs and MRIs simultaneously, and based on those patterns, predict characteristics of a patient’s heart. Such a tool, with further development, could one day help doctors better detect and diagnose heart conditions from routine tests such as ECGs.
The researchers also showed that they could analyze ECG recordings, which are easy and cheap to acquire, and generate MRI movies of the same heart, which are much more expensive to capture. And their method could even be used to find new genetic markers of heart disease that existing approaches that look at individual data modalities might miss.
Overall, the team said their technology is a more holistic way to study the heart and its ailments. “It is clear that these two views, ECGs and MRIs, should be integrated because they provide different perspectives on the state of the heart,” said Caroline Uhler, a co-senior author on the study, a Broad core institute member, co-director of the Schmidt Center at Broad, and a professor in the Department of Electrical Engineering and Computer Science as well as the Institute for Data, Systems, and Society at MIT.
"As a field, cardiology is fortunate to have many diagnostic modalities, each providing a different view into cardiac physiology in health and diseases. A challenge we face is that we lack systematic tools for integrating these modalities into a single, coherent picture,” said Anthony Philippakis, a senior co-author on the study and chief data officer at Broad and co-director of the Schmidt Center. “This study represents a first step towards building such a multi-modal characterization."
Model making
To develop their model, the researchers used a machine learning algorithm called an autoencoder, which automatically integrates gigantic swaths of data into a concise representation – a simpler form of the data. The team then used this representation as input for other machine learning models that make specific predictions.
In their study, the team first trained their autoencoder using ECGs and heart MRIs from participants in the UK Biobank. They fed in tens of thousands of ECGs, each paired with MRI images from the same person. The algorithm then created shared representations that captured crucial details from both types of data.
“Once you have these representations, you can use them for many different applications,” said Adityanarayanan Radhakrishnan, a co-first author on the study, an Eric and Wendy Schmidt Center Fellow at the Broad, and a graduate student at MIT in Uhler’s lab. Sam Friedman, a senior machine learning scientist in the Data Sciences Platform at the Broad, is the other co-first author.
One of those applications is predicting heart-related traits. The researchers used the representations created by their autoencoders to build a model that could predict a range of traits, including features of the heart like the weight of the left ventricle, other patient characteristics related to heart function like age, and even heart disorders. Moreover, their model outperformed more standard machine learning approaches, as well as autoencoder algorithms that were trained on just one of the imaging modalities.
“What we showed here is that you get better prediction accuracy if you incorporate multiple types of data,” Uhler said.
Radhakrishnan explained that their model made more accurate predictions because it used representations that had been trained on a much larger dataset. Autoencoders don’t require data that have been labeled by humans, so the team could feed their autoencoder with around 39,000 unlabeled pairs of ECGs and MRI images, rather than just around 5,000 labeled pairs.
The researchers demonstrated another application of their autoencoder: generating new MRI movies. By inputting an individual’s ECG recording into the model — without a paired MRI recording — the model produced the predicted MRI movie for the same person.
With more work, the scientists envision that such technology could potentially allow physicians to learn more about a patient’s heart health from just ECG recordings, which are routinely collected at doctors’ offices.
Broader gene search
With their autoencoder representations, the team realized they could also use them to look for genetic variants associated with heart disease. The traditional method of finding genetic variants for a disease, called a genome-wide association study (GWAS), requires genetic data from individuals that have been labeled with the disease of interest.
But because the team’s autoencoder framework doesn’t require labeled data, they were able to generate representations that reflected the overall state of a patient’s heart. Using these representations and genetic data on the same patients from the UK Biobank, the researchers created a model that looked for genetic variants that impact the state of the heart in more general ways. The model produced a list of variants including many of the known variants related to heart disease and some new ones that can now be investigated further.
Radhakrishnan said that genetic discovery could be the area in which the autoencoder framework, with more data and development, could have the most impact – not just for heart disease, but for any disease. The research team is already working on applying their autoencoder framework to study neurological diseases.
Uhler said this project is a good example of how innovations in biomedical data analysis emerge when machine learning researchers collaborate with biologists and physicians. “An exciting aspect about getting machine learning researchers interested in biomedical questions is that they might come up with a completely new way of looking at a problem.”
Support for the research was provided in part by the Eric and Wendy Schmidt Center at the Broad Institute, the National Science Foundation, the Office of Naval Research, the MIT-IBM Watson AI Lab, a Simons Investigator Award, the National Institutes of Health, and the American Heart Association.
Adapted from a news story posted on the Broad Institute website.
In a recent grant application to the National Institutes of Health, Petar Stojanov was required, among other things, to describe his “specific aims” as well as his background. It’s doubtful that the NIH reviewers would have considered Stojanov’s research agenda lacking in ambition, given its broad scope: to identify the genetic mutations that cause cancer and figure out how they cause it.
The reviewers, moreover, must have decided he had a credible chance of achieving these goals, or at least making progress toward their realization, as he was informed earlier this year that he had earned a coveted Pathway to Independence (K99) Award. As a result, Stojanov — a current Eric and Wendy Schmidt Center Postdoctoral Fellow at the Broad Institute of MIT and Harvard — will receive up to five years of research support, meaning he can devote himself fully to his scientific inquiries without having to worry about funding.
K99 grants help “outstanding” researchers transition from postdoctoral positions to running their own labs. In this next stage of his career, Stojanov will develop new methods in two types of machine learning: algorithms related to causality and deep generative models.
An early interest in computational biology
In some sense, Stojanov set off on the path that led him to this milestone when he was a high school student in Macedonia. A family friend told him that computational biology was becoming a hot area in science. Stojanov was immediately intrigued, he said, “for the same reason that has brought many people to this field — math and biology were my favorite subjects.” And here was a chance to combine his preferred disciplines into a unified course of study that might lead to an interesting career.
He spent his senior year of high school in Pelham, New York (where he lived with his family friend), as he’d always believed he “would have the best opportunities for innovation in the U.S.” A year later, he enrolled in Bard College, which had no courses, let alone a major, in computational biology. Stojanov stuck to his passion, nevertheless, taking the bulk of his classes in computer science, biology, mathematics, and chemistry. He gained hands-on experience in computational biology through summer research programs at George Washington University and the University of Maryland.
Stojanov on his way to work at the Broad Institute
After graduating from Bard in 2010, he took a job in the laboratory of Gaddy Getz, director of the Broad’s Cancer Genome Computational Analysis Group. That’s where Stojanov got started on the two-pronged research track he’s still pursuing today: First, to figure out which mutations are present in cancerous tissue and, second, to determine which of those mutations actually spur our cells to multiply out of control and drive cancer. The standard approach at the time was to rely on statistical methodology, such as examining whether the number of mutations in a given gene was greater than would be expected from random processes, unrelated to cancer.
Stojanov spent four productive years at the Broad, coauthoring more than a dozen papers — four of which he was a lead author. He didn’t sleep much those days, mainly because he was “hungry for projects and never said no to an opportunity.” Yet, by the end of that tenure, he felt that his work in this area could benefit from additional training in computer science, which would enable him to bring new tools to the kinds of problems he’d been grappling with. In 2014, he entered a PhD program at Carnegie Mellon University, where he immersed himself in machine learning techniques and other emerging approaches in artificial intelligence. Although his graduate research had nothing to do with biology, he recognized that the methods he was learning, combined with statistics, might lead to breakthroughs in his previous cancer investigations.
Bringing ML to bear on cancer research
Stojanov returned to the Broad in 2021 and picked up in the Getz lab where he had left off — this time ready to unleash the full power of AI. Getz was eager to have him back, touting “the unique set of skills that Petar has,” given his prior experience in cancer research and his recently strengthened background in computer science. “And now,” Getz said, “he’s applying his expertise in machine learning to the search for the drivers of cancer.”
Just counting the number of mutations in a gene is not enough to reveal the mechanisms underpinning cancer, Stojanov explained. “That may tell you which mutations are most prevalent, and maybe the most important, but it still doesn’t tell you what they do.” To understand how a mutation affects a gene, you have to look at gene expression, the cellular process by which the information encoded in a gene is used to create proteins.
In his latest work at the Broad, Stojanov is focusing on two variables: gene mutations, which can be gleaned from DNA sequencing data, and gene expression expression (which can be obtained from RNA sequencing data by measuring the amount of RNA, a gene-decoding molecule, in the cell). He then uses a set of machine learning tools called causal inference and discoveryalgorithms to uncover the “causal relationships” between these two variables – mutations and expression.
“The idea is to show that some aspects of gene expression are the consequences of mutations,” he said.
The only causal relationships he cares about are those associated with cancer. While sorting through DNA and RNA sequencing data from thousands of cancer patients, he’s looking for patterns. In particular, he said, “we might find mutations that influence patients with the same cancer type (or subtype), in the same way.”
Stojanov in his office with colleagues Pinar Eser (center) and Tim Coorens
As an intermediate step, Stojanov relies on a related class of machine learning-based tools, so-called deep generative models, which basically takes abstract (“high-dimensional”) information processed by computers and represents it in a form that is meaningful to humans. If you have mutation and expression data for 20,000 genes, he said, these models offer a way to summarize that vast amount of data in terms of the concepts you’re interested in, such as biological processes or cell subtypes that might be impacted by cancer.
The ultimate goal is to learn as much as possible about this multifaceted disease — how and where it starts and progresses. “To really understand what’s going on,” Stojanov said, “we need an interpretable map that shows which processes are affected by what mutations.”
Existing techniques can only get you so far
Eric and Wendy Schmidt Center co-director Caroline Uhler is excited by the prospect of “getting at the causal genes, which contain the mutations that drive cancer. "Once you have that,” she said, “you’re in a much better position to think about effective therapies. That’s really the promise of this work.”
Stojanov’s current research is, admittedly, at an early stage. He has a solid base of experience to draw on, and he’s picked out a set of tools, in the form of machine learning algorithms, that are poised to advance our knowledge base. The big challenge, Uhler pointed out, is that “existing techniques can only get you so far. Petar has to build on these methods and develop new algorithms in order to solve the important biological questions he plans to address.”
Stojanov is mindful of the hard work ahead and grateful that his burden has been eased by having several years of funding already secured. “This [K99] award gives you the ultimate amount of independence you can have as a postdoc,” he said.
When asked if getting the award is the best thing that could happen to someone in his position, embarking on such an ambitious enterprise, he replied, “Well, it’s certainly up there.”
Neural networks, a type of machine-learning model, are being used to help humans complete a wide variety of tasks, from predicting if someone’s credit score is high enough to qualify for a loan to diagnosing whether a patient has a certain disease. But researchers still have only a limited understanding of how these models work. Whether a given model is optimal for certain task remains an open question.
MIT researchers have found some answers. They conducted an analysis of neural networks and proved that they can be designed so they are “optimal,” meaning they minimize the probability of misclassifying borrowers or patients into the wrong category when the networks are given a lot of labeled training data. To achieve optimality, these networks must be built with a specific architecture.
The researchers discovered that, in certain situations, the building blocks that enable a neural network to be optimal are not the ones developers use in practice. These optimal building blocks, derived through the new analysis, are unconventional and haven’t been considered before, the researchers say.
In a paper published this week in the Proceedings of the National Academy of Sciences, they describe these optimal building blocks, called activation functions, and show how they can be used to design neural networks that achieve better performance on any dataset. The results hold even as the neural networks grow very large. This work could help developers select the correct activation function, enabling them to build neural networks that classify data more accurately in a wide range of application areas, explains senior author Caroline Uhler, a professor in the Department of Electrical Engineering and Computer Science (EECS) and co-director of the Eric and Wendy Schmidt Center at the Broad Institute of MIT and Harvard.
“While these are new activation functions that have never been used before, they are simple functions that someone could actually implement for a particular problem. This work really shows the importance of having theoretical proofs. If you go after a principled understanding of these models, that can actually lead you to new activation functions that you would otherwise never have thought of,” says Uhler, who is a core institute member of the Broad Institute, and a researcher at MIT’s Laboratory for Information and Decision Systems (LIDS) and Institute for Data, Systems and Society (IDSS).
Joining Uhler on the paper are lead author Adityanarayanan Radhakrishnan, an EECS graduate student and an Eric and Wendy Schmidt Center Fellow, and Mikhail Belkin, a professor in the Halicioğlu Data Science Institute at the University of California at San Diego.
Activation investigation
A neural network is a type of machine-learning model that is loosely based on the human brain. Many layers of interconnected nodes, or neurons, process data. Researchers train a network to complete a task by showing it millions of examples from a dataset.
For instance, a network that has been trained to classify images into categories, say dogs and cats, is given an image that has been encoded as numbers. The network performs a series of complex multiplication operations, layer by layer, until the result is just one number. If that number is positive, the network classifies the image a dog, and if it is negative, a cat.
Activation functions help the network learn complex patterns in the input data. They do this by applying a transformation to the output of one layer before data are sent to the next layer. When researchers build a neural network, they select one activation function to use. They also choose the width of the network (how many neurons are in each layer) and the depth (how many layers are in the network.)
“It turns out that, if you take the standard activation functions that people use in practice, and keep increasing the depth of the network, it gives you really terrible performance. We show that if you design with different activation functions, as you get more data, your network will get better and better,” says Radhakrishnan.
He and his collaborators studied a situation in which a neural network is infinitely deep and wide — which means the network is built by continually adding more layers and more nodes — and is trained to perform classification tasks. In classification, the network learns to place data inputs into separate categories.
“A clean picture”
After conducting a detailed analysis, the researchers determined that there are only three ways this kind of network can learn to classify inputs. One method classifies an input based on the majority of inputs in the training data; if there are more dogs than cats, it will decide every new input is a dog. Another method classifies by choosing the label (dog or cat) of the training data point that most resembles the new input.
The third method classifies a new input based on a weighted average of all the training data points that are similar to it. Their analysis shows that this is the only method of the three that leads to optimal performance. They identified a set of activation functions that always use this optimal classification method.
“That was one of the most surprising things — no matter what you choose for an activation function, it is just going to be one of these three classifiers. We have formulas that will tell you explicitly which of these three it is going to be. It is a very clean picture,” he says.
They tested this theory on a several classification benchmarking tasks and found that it led to improved performance in many cases. Neural network builders could use their formulas to select an activation function that yields improved classification performance, Radhakrishnan says.
In the future, the researchers want to use what they’ve learned to analyze situations where they have a limited amount of data and for networks that are not infinitely wide or deep. They also want to apply this analysis to situations where data do not have labels.
“In deep learning, we want to build theoretically grounded models so we can reliably deploy them in some mission-critical setting. This is a promising approach at getting toward something like that — building architectures in a theoretically grounded way that translates into better results in practice,” he says.
This work was supported, in part, by the National Science Foundation, Office of Naval Research, the MIT-IBM Watson AI Lab, the Eric and Wendy Schmidt Center at the Broad Institute, and a Simons Investigator Award.
Marios Gavrielatos had never participated in a machine learning competition when he decided to enter the Eric and Wendy Schmidt Center’s Cancer Immunotherapy Data Science Grand Challenge.
Gavrielatos’ friend and colleague, Konstantinos Kyriakidis, asked him to team up in the competition after learning about it from a promotional video on YouTube.
Despite Gavrielatos’ newcomer status, the pair developed a new deep learning model that won them the first part of the competition last month.
The challenge “helped me develop new computational skills, deep-learning wise,” said Gavrielatos, a bioinformatics master’s student at the National and Kapodistrian University of Athens, adding that because they couldn’t find similar problems online, “we had to develop something new ourselves, which was interesting.”
The Cancer Immunotherapy Data Science Grand Challenge, which ran on Topcoder from January 9 to February 3, aimed to uncover new ways to modify, or “perturb,” T cells to make them more effective at killing cancer cells to ultimately improve cancer treatment.
Top challenge submissions will be tested out in a lab at the Broad Institute of MIT and Harvard later this year.
The Eric and Wendy Schmidt Center at the Broad Institute of MIT and Harvard partnered with Harvard’s Laboratory for Innovation Science, the MIT Department of Electrical Engineering and Computer Science, Topcoder, Gordian Biotechnology, and Massachusetts General Hospital (MGH) to run the challenge. Over 900 people registered for the first part of the competition — making it Topcoder’s fifth-largest data science challenge to date.
“In biology, we can perform perturbations on a scale that other fields can only dream of, meaning we need to develop novel machine learning methods to best make use of such data and answer biological questions,” said Caroline Uhler, co-director of the Eric and Wendy Schmidt Center, a core member of the Broad Institute, and professor in the Department of Electrical Engineering and Computer Science and the Institute for Data, Systems and Society at MIT. “We held this data science challenge to direct bright computational minds from around the world to this problem in cancer immunotherapy. And we’re thrilled that we now get to test out some of their proposed perturbations experimentally.”
A great fit for a data science challenge
While chemotherapy and radiation have saved many lives, these treatments have a weak spot: they are not specific enough — meaning they can kill cancerous and healthy cells. The promise of cancer immunotherapy, a newer and effective form of cancer treatment, is that it can harness our immune system to recognize and kill cancer cells while leaving other cells alone in most cases.
Cancer cells have developed a number of ways to evade our immune system. One such strategy is sending signals to T cells to make them exhausted and ineffective at killing cancer cells. That’s why cancer researchers like Nir Hacohen, an institute member at Broad, director of the Broad Institute’s Cell Circuits Program and director of the Center for Cancer Immunology at Mass General Hospital, are investigating whether perturbing certain genes could shift T cells to a cancer-fighting, “effector” state.
“We were excited to develop this data science challenge with the Eric and Wendy Schmidt Center because the T cell exhaustion problem seemed like a great fit for this kind of competition,” said Hacohen. “It was an opportunity to combine our cancer biology and immunology knowledge with the computational and mathematical skills of machine learning experts from all over the world.”
Marc Schwartz, a postdoctoral fellow in the Hacohen Lab, ran experiments testing the effects of 73 gene knockouts in T cells on mice with cancer. Given that it took months to test a fraction of the 20,000 potential gene knockouts — a genetic perturbation that stops a gene from functioning — Broad researchers wanted a way to zero in on the most promising perturbations. Enter machine learning.
The overarching challenge was divided into three parts that ran as individual data science competitions on Topcoder. In Challenge 1, participants received gene expression data from 66 of the 73 T-cell-gene knockouts from Schwartz’s experiments as training data. They then had to develop an algorithm that could predict how knocking out the seven “held-out” genes would affect T cells.
Challenge 2 participants used their algorithms from the first challenge to propose new gene knockouts (picking from any of the 20K genes in the entire genome) to shift as many T cells as possible into a cancer-fighting state. In Challenge 3, participants proposed a metric for ranking how well a particular gene knockout would bring about this desired shift in T cells.
To solve Challenge 1, winners Gavrielatos and Kyriakidis first pared down the single-cell dataset so that it contained only expression information from important genes — that is, genes whose expression changed across different T cell states. The preprocessing of the data is a crucial step to distill the “signal” — or useful information — when working with such noisy data, said Kyriakidis, who has previously won several precision FDA data science challenges.
The pair next trained a deep learning model to predict what portion of T cells would move into an effector, exhausted, or alternate state after a specific gene was knocked out. Initially, they tried to come up with an algorithm using only the training data provided from Schwartz’s experiment. But as they continued working, they realized that incorporating public biomedical databases into their analysis — namely, Reactome, a database of biological pathways in human cells, and STRING, a protein interaction database — could reveal associations between the missing and observed genes.
“The whole process was so rewarding,” said Kyriakidis. “You have to divide the whole problem into smaller parts to try to find the solution to each part and connect the dots.”
Sometimes, simple algorithms are best
The second place winners were three MIT students — including two graduate students from the Laboratory for Information and Decision Systems (LIDS), Yuzhou Gu and Anzo Teh, MIT Institute for Data, Systems, and Society (IDSS) postdoc Yanjun Han, and undergraduate student Brandon Wang. Teh, who is also an Eric and Wendy Schmidt Center PhD fellow, said his advisor, MIT professor Yury Polyanskiy, suggested that he and the other researchers join forces for the challenge.
Anzo Teh, Eric and Wendy Schmidt Center PhD Fellow
Teh, Gu, and Han, have a theoretical and computational background — specifically, information theory — while the undergraduate student, Brandon Wang, has expertise in computational biology.
“I did feel like this challenge was a good way for me to learn how to work on these types of problems because I’m pretty new to the biology field,” said Teh.
Several teams used neural networks to describe the experimental gene expression data, an approach that often requires thousands of parameters to create an effective model. The MIT team, on the other hand, made a simplifying assumption that gene expression could be modeled with a small number of parameters following a Gaussian distribution, or a bell curve.
They then reduced the dimensions of their data from 20,000 to 50 columns using a machine learning technique called “principal component analysis.” The MIT team also incorporated an outside public database on human genes into their model, mapping human gene expression profiles to their missing mouse counterparts. Finally, they used a proven machine learning classification algorithm to determine how the gene expression profiles lined up with T cell states.
“Sometimes simple algorithms can work better than neural networks,” said Teh. The MIT team’s background in information theory, which is the study of organizing and quantifying data, helped them discover what signals in the experimental data to focus their models on.
Peter Novotný, the third place winner and a math professor at the University of Žilina in Slovakia, also took a relatively simple approach to solving Challenge 1. Novotný, a former Topcoder “copilot” who had participated in a NASA asteroid-hunter challenge, among many other competitions, has more of a mathematics than a computer science background. In part through participating in data science challenges, he’s discovered that he enjoys machine learning though.
“And, I also quite like competing,” he said.
For the cancer immunotherapy challenge, Novotný first selected 14 features from the T cell data that quantified how gene expression levels differed between perturbed and unperturbed cells, as the way to represent his training data. Then, he built a model using a common machine learning algorithm — the “random forest” — and predicted the distribution of T cell states for each of the seven withheld genes.
To make the challenge accessible to participants without a biology background, Lightmark Creative and Orr Ashenberg, associate director of computational biology at The Klarman Cell Observatory of the Broad Institute, produced a 1.5-hour crash course on cancer biology, perturbation data, and single-cell sequencing technologies.
“To compete in this contest, you really need to understand what the data is, and without those lectures, it would be quite difficult to understand the problem,” said Novotný.
In addition, Uhler held an IAP course that ran at the same time as the challenge, encouraging MIT students to team up and participate in the competition.
Testing perturbations in the lab
The Eric and Wendy Schmidt Center also announced last month who won the third challenge, in which participants came up with a metric to rank new T cell perturbations.
The winners of that challenge were:
First place: Dariusz Brzeziński and Wojciech Kotlowski from Poznań University of Technology in Poland
Second place: Salil Bhate, MIT, postdoctoral fellow at the Eric and Wendy Schmidt Center
Third place: Irene Bonafonte Pardàs, Artur Szalata, and Benjamin Schubert from Helmholtz Center Munich and Miriam Lyzotte from Mila - Quebec AI Institute
Now, researchers at the Hacohen Lab will run experiments to test how the perturbations proposed in Challenge 2 affect mouse T cells’ cancer-fighting abilities.
“It will be really exciting to see how these computationally identified perturbations actually perform in the lab,” said Uhler. “After all, machine learning cannot replace experiments, but the goal is to work hand in hand with biologists and help prioritize the next experiments to run.”
The name Sybil has its origins in the oracles of Ancient Greece, also known as sibyls: feminine figures who were relied upon to relay divine knowledge of the unseen and the omnipotent past, present, and future. Now, the name has been excavated from antiquity and bestowed on an artificial intelligence tool for lung cancer risk assessment being developed by researchers at MIT's Abdul Latif Jameel Clinic for Machine Learning in Health, Mass General Cancer Center (MGCC), and Chang Gung Memorial Hospital (CGMH).
Lung cancer is the No. 1 deadliest cancer in the world, resulting in 1.7 million deaths worldwide in 2020, killing more people than the next three deadliest cancers combined.
"It’s the biggest cancer killer because it’s relatively common and relatively hard to treat, especially once it has reached an advanced stage,” says Florian Fintelmann, MGCC thoracic interventional radiologist and co-author on the new work. “In this case, it’s important to know that if you detect lung cancer early, the long-term outcome is significantly better. Your five-year survival rate is closer to 70 percent, whereas if you detect it when it’s advanced, the five-year survival rate is just short of 10 percent.”
Although there has been a surge in new therapies introduced to combat lung cancer in recent years, the majority of patients with lung cancer still succumb to the disease. Low-dose computed tomography (LDCT) scans of the lung are currently the most common way patients are screened for lung cancer with the hope of finding it in the earliest stages, when it can still be surgically removed. Sybil takes the screening a step further, analyzing the LDCT image data without the assistance of a radiologist to predict the risk of a patient developing a future lung cancer within six years.
In their new paper published in the Journal of Clinical Oncology, Jameel Clinic, MGCC, and CGMH researchers demonstrated that Sybil obtained C-indices of 0.75, 0.81, and 0.80 over the course of six years from diverse sets of lung LDCT scans taken from the National Lung Cancer Screening Trial (NLST), Mass General Hospital (MGH), and CGMH, respectively — models achieving a C-index score over 0.7 are considered good and over 0.8 is considered strong. The ROC-AUCs for one-year prediction using Sybil scored even higher, ranging from 0.86 to 0.94, with 1.00 being the highest score possible.
Despite its success, the 3D nature of lung CT scans made Sybil a challenge to build. Co-author Peter Mikhael, an MIT PhD student in electrical engineering and computer science, a fellow at the Eric and Wendy Schmidt Center, and an affiliate at the Jameel Clinic and the MIT Computer Science and Artificial Intelligence Laboratory (CSAIL), likened the process to “trying to find a needle in a haystack.” The imaging data used to train Sybil was largely absent of any signs of cancer because early-stage lung cancer occupies small portions of the lung — just a fraction of the hundreds of thousands of pixels making up each CT scan. Denser portions of lung tissue are known as lung nodules, and while they have the potential to be cancerous, most are not, and can occur from healed infections or airborne irritants.
To ensure that Sybil would be able to accurately assess cancer risk, Fintelmann and his team labeled hundreds of CT scans with visible cancerous tumors that would be used to train Sybil before testing the model on CT scans without discernible signs of cancer.
MIT electrical engineering and computer science PhD student Jeremy Wohlwend, co-author of the paper and Jameel Clinic and CSAIL affiliate, was surprised by how highly Sybil scored despite the lack of any visible cancer. “We found that while we [as humans] couldn’t quite see where the cancer was, the model could still have some predictive power as to which lung would eventually develop cancer,” he recalls. “Knowing [Sybil] was able to highlight which side was the most likely side was really interesting to us.”
Co-author Lecia V. Sequist, a medical oncologist, lung cancer expert, and director of the Center for Innovation in Early Cancer Detection at MGH, says the results the team achieved with Sybil are important “because lung cancer screening is not being deployed to its fullest potential in the U.S. or globally, and Sybil may be able to help us bridge this gap.”
Lung cancer screening programs are underdeveloped in regions of the United States hardest hit by lung cancer due to a variety of factors. These range from stigma against smokers to political and policy landscape factors like Medicaid expansion, which varies from state to state.
Moreover, many patients diagnosed with lung cancer today have either never smoked or are former smokers who quit over 15 ago — traits that make both groups ineligible for lung cancer CT screening in the United States.
“Our training data consisted only of smokers because this was a necessary criterion for enrolling in the NLST,” Mikhael says. “In Taiwan, they screen nonsmokers, so our validation data is expected to contain people who didn’t smoke, and it was exciting to see Sybil generalize well to that population.”
“An exciting next step in the research will be testing Sybil prospectively on people at risk for lung cancer who have not smoked or who quit decades ago,” says Sequist. “I treat such patients every day in my lung cancer clinic and it’s understandably hard for them to reconcile that they would not have been candidates to undergo screening. Perhaps that will change in the future.”
There is a growing population of patients with lung cancer who are categorized as nonsmokers. Women nonsmokers are more likely to be diagnosed with lung cancer than men who are nonsmokers. Globally, over 50 percent of women diagnosed with lung cancer are nonsmokers, compared to 15 to 20 percent of men.
MIT Professor Regina Barzilay, a paper co-author and the Jameel Clinic AI faculty lead, who is also a member of the Koch Institute for Integrative Cancer Research, credits MIT and MGH’s joint efforts on Sybil to Sylvia, the sister to a close friend of Barzilay and one of Sequist’s patients. "Sylvia was young, healthy and athletic — she never smoked,” Barzilay recalls. “When she started coughing, neither her doctors nor her family initially suspected that the cause could be lung cancer. When Sylvia was finally diagnosed and met Dr. Sequist, the disease was too advanced to revert its course. When mourning Sylvia's death, we couldn't stop thinking how many other patients have similar trajectories.”
This work was supported by the Bridge Project, a partnership between the Koch Institute at MIT and the Dana-Farber/Harvard Cancer Center; the MIT Jameel Clinic; Quanta Computer; Stand Up To Cancer; the MGH Center for Innovation in Early Cancer Detection; the Bralower and Landry Families; Upstage Lung Cancer; and the Eric and Wendy Schmidt Center at the Broad Institute of MIT and Harvard. The Cancer Center of Linkou CGMH under Chang Gung Medical Foundation provided assistance with data collection and R. Yang, J. Song and their team (Quanta Computer Inc.) provided technical and computing support for analyzing the CGMH dataset. The authors thank the National Cancer Institute for access to NCI’s data collected by the National Lung Screening Trial, as well as patients who participated in the trial.
Many diseases affect how cells are spatially organized in tissues, such as in Alzheimer’s disease, where amyloid-β proteins clump together to form plaques in the brain. Studying how cells differ in various regions of tissue could help scientists better understand the key changes that lead to Alzheimer’s and other diseases. But integrating data on gene expression and cell structure and spatial location into the same analysis has proven challenging.
Now, researchers from the Broad Institute of MIT and Harvard and ETH Zürich in Switzerland have developed a computational framework for simultaneously analyzing gene expression, the structure of cell nuclei, and their position in space. STACI (Spatial Transcriptomics combined using Autoencoders with Chromatin Imaging) is the first method that combines these three kinds of data. The findings appeared recently in Nature Communications.
The team, led by Caroline Uhler, the study’s senior author and co-director of the Eric and Wendy Schmidt Center at the Broad, and Xinyi Zhang, first author on the study and a graduate student in Uhler’s lab, developed STACI and applied it to study a mouse model of Alzheimer’s disease.
STACI uses a kind of computational model called a neural network to analyze data generated by a technique called STARmap, which measures the expression of more than two thousand genes and maps their location in intact tissue. STARmap was developed by Xiao Wang, a core institute member at the Broad and co-author on the study.
The team used STACI to analyze brain tissue from the Alzheimer’s mouse model. By studying gene expression and the location of cells in the tissue, the scientists identified a part of the cortex in the mouse brain that was more likely to have significant plaque accumulations. With the help of G. V. Shivashankar, a study author and professor of mechano-genomics at ETH Zürich, the team also found that they could predict plaque size — a marker of disease progression — by analyzing just one feature of cells near the plaques: the structure of chromatin, the complex of DNA and protein that makes up chromosomes. The results suggest that chromatin structure could be a marker of Alzheimer’s disease progression.
“We began by asking how we can integrate these different data modalities,” said Uhler, who is also a core institute member at Broad and professor in the Department of Electrical Engineering and Computer Science at MIT. “What’s really exciting is that now, with STACI, we can begin to ask biological questions to learn more about disease by taking all modalities into account simultaneously.”
Zhang, who is also a fellow at the Schmidt Center, says that STACI is a useful tool for researchers because chromatin imaging is routine in labs and cheaper than measuring the gene expression of cells directly. “This study may provide simple, low-cost avenues for studying which regions of the brain are more affected by disease and for tracking disease progression,” she said.
Cells in space
In previous work, Uhler and Shivashankar showed that they could use computational techniques to analyze single-cell RNA sequencing data along with chromatin images. They collaborated with Wang to incorporate the analysis of cell location data from STARmap and build STACI.
STACI relies on a neural network, which learns patterns from “training” data to predict characteristics of new data. To develop STACI, the researchers trained it to build a map, called a latent space, that groups together cells with similar locations, gene expression, or chromatin structure. They then used STACI to analyze images of chromatin from mouse brain tissue.
From this latent space, the scientists found that the size of plaque deposits is highly correlated with the ratio of heterochromatin to euchromatin, which indicates how densely packed the chromatin is. This relationship suggests DNA packing could be a marker of disease progression.
The team says the connection between chromatin density and plaques suggests new questions in Alzheimer’s research. They hope their findings will spur other groups to investigate the biological relationship between DNA packing and plaque build-up.
Branching out
Brain tissue samples can vary widely in how they are collected and prepared, but the scientists designed STACI to account for this variation. The technique could also be applicable to other spatial data types, such as from Slide-Seq — developed by Fei Chen, Evan Macosko and other colleagues at the Broad — as well as Visium and MERFISH.
Uhler adds that STACI could also help researchers learn more about other diseases, since many have important spatial features. She envisions using the framework to analyze the local microenvironment in cancer, fibrosis or scarring in the lungs or other tissues, as well as developmental processes. As scientists apply STACI to new problems, they’ll likely encounter new analytical challenges, but she thinks this is an opportunity to help the model expand.
“This work shows how biology can be a great inspiration for novel computational questions and developments,” Uhler said. “And that’s really exciting.”
This work was supported in part by the Eric and Wendy Schmidt Center, the Simons Foundation, the Office of Naval Research, the National Institutes of Health, and the National Science Foundation.
Adapted from a news story posted on the Broad Institute website.
The immune system is adept at fighting off viral and bacterial infections, but it can also find and attack cancer in the body. Cancer cells, however, are skilled at disarming the immune system’s T cells — allowing tumors to continue growing unabated.
Scientists at the Broad Institute of MIT and Harvard and beyond have been looking for ways to genetically modify T cells to improve their cancer-fighting ability. Now the Eric and Wendy Schmidt Center at the Broad Institute is joining this effort, by holding a data science challenge this winter that will call on machine learning enthusiasts to develop algorithms that identify effective genetic modifications in T cells.
Winners will receive monetary prizes at each stage — and, unlike in most data science challenges, the top-scoring participants will have their submissions experimentally validated. Members of a cancer immunology lab at Broad led by institute member Nir Hacohen will make the top-ranked genetic modifications in T cells in the lab and assess the cells’ cancer-fighting abilities.
The "Cancer Immunotherapy Data Science Grand Challenge" was announced earlier this month at the online coding tournament Topcoder Open, and will run from January 9 to February 3, 2023. The Eric and Wendy Schmidt Center is partnering with Harvard’s Laboratory for Innovation Science, the MIT Department of Electrical Engineering and Computer Science, Topcoder, and Massachusetts General Hospital (MGH) to run the challenge.
“Machine learning experts have largely gone into the fields of big technology and finance. With this challenge, we’re describing an important problem in cancer immunology in a way that is approachable for computational minds — thus hoping to entice more of these experts to the life sciences,” said Caroline Uhler, co-director of the Eric and Wendy Schmidt Center, a core member of the Broad Institute, and professor in the Department of Electrical Engineering and Computer Science and the Institute for Data, Systems and Society at MIT.
Improving cancer immunotherapy through machine learning
Cancer immunotherapies boost the immune system to fight off cancer in a variety of ways. Scientists have made many breakthroughs in cancer immunotherapy in the last decade, such as the development of several FDA-approved checkpoint blockade and “CAR T” therapies. CAR T treatments involve removing T cells from a cancer patient, genetically engineering them in the lab to target tumors, and then reintroducing them back into the patient. However, these treatments work for only a small number of cancer types and only in some patients.
“We hope that this challenge will allow us to quickly hone in on the most promising perturbations so we can better target our experimental validation.” — Nir Hacohen
To make T cell-based immunotherapies more effective for more patients, scientists are looking for other genetic changes they can introduce in T cells to make them better cancer killers. With the development of genome-editing technologies such as CRISPR in the last decade, researchers can look for those desirable changes by performing large-scale genetic screens to systematically modify or knock out each gene and study the effect of these “perturbations” at the single-cell level.
However, perturbing each of the 20,000 genes in the cell or the several hundred million different combinations of genes in the lab would be too costly and time-consuming. Machine learning can help, by predicting which genetic perturbations might be most effective.
“We hope that this challenge will allow us to quickly hone in on the most promising perturbations so we can better target our experimental validation,” said Hacohen, director of the Broad Institute’s Cell Circuits Program, institute member of the Broad Institute, and director of MGH’s Center for Cancer Immunology. “The predictions from this challenge will provide a crucial step toward making cancer immunotherapy more effective for more patients.”
The Cancer Immunotherapy Data Science Challenge will consist of three parts that will run at the same time. In the first part, participants will use transcriptomic and perturbational data from T cells in mouse tumors to develop algorithms that predict the effect of perturbations that have already been studied in the lab, allowing them to see how well their algorithms work. In part two, they’ll come up with a metric for ranking how well a particular gene knockout would shift T cells to a desired state.
And, third, participants will use their algorithms to propose perturbations that boost T cells’ ability to destroy tumors. The top-scoring participants from part one will have their proposed perturbations experimentally validated.
“Data science challenges like this one draw on the power of the crowd to bring in outside computational and creative machine learning techniques to solve biological problems,” said MarcAntonio Awada, head of research and data science at Harvard’s Institute for Digital, Data, and Design Institute. “In the past, crowdsourcing has led to out-of-the-box approaches and completely novel solutions compared to what experts had come up with.”
Unique learning and data access opportunities
The challenge will run concurrently with an Independent Activities Period course at MIT, which brings together computer science and biology students to collaborate on this problem. “The course provides a great opportunity for MIT students to apply their education and see that what they’re learning in the classroom has a direct impact on answering critical biomedical questions,” said Uhler, who is one of the course’s instructors.
A biology background isn’t necessary to participate. The Eric and Wendy Schmidt Center will provide all challenge participants with an online crash course on cancer immunology and unique features of the large-scale datasets. Interested participants can pre-register now as an individual or as part of a team on Topcoder, which is hosting the challenge on their platform.
Participants will have free access to Saturn Cloud to complete the challenge.
Adapted from a news story posted on the Broad Institute website.
Advancing our understanding of tissue biology requires tight collaborations between biologists with driving questions, technologists creating new experimental methods, and computational scientists who are creating new ways of analyzing data. One of the key aims of an April 27 workshop held by the Eric and Wendy Schmidt Center and the Klarman Cell Observatory at the Broad Institute was to explore the interface between these disciplines. Speakers and panelists included researchers at Stanford University, MIT, Harvard University, the Sloan Kettering Institute, UC Berkeley, Princeton University, and the Broad Institute.
The workshop brought together a diverse set of communities to discuss new tissue biology research questions — and new opportunities for collaboration between the biomedical sciences and machine learning.
Caroline Uhler, co-director of the Eric and Wendy Schmidt Center, a core member of the Broad Institute, and an associate professor in the Department of Electrical Engineering and Computer Science and the Institute for Data, Systems and Society at MIT, told workshop attendees during opening remarks that biology has seen an “explosion” of data in recent years. “We now have the opportunity to understand the programs of life, so not just the units (like genes or single cells), but actually the interactions between these units.”
Biological research frontiers
These cellular interactions play a key role in the cancer immunotherapy research shared by keynote speaker Garry Nolan, a professor in the pathology department at Stanford University. His research team develops algorithms to model tissue areas where different groups of cells interact, areas he calls “interface zones,” to gain insights into how cancer remodels its surrounding tissue and evades the body’s immune system. These interface zones are critical as the locus of cellular changes that lead to tumor growth.
“I would urge you, when you're looking at your RNA data sets, to the extent that you can call out these kinds of interface zones, pay special attention to the RNA changes that are occurring there,” said Nolan, adding later: “The boundary space is where the action is.”
Additionally, biologists should reconsider labeling tumors and other features “heterogenous,” which implies that tumors from different patients are too distinct from one another to be compared. “There is an order here that can be extracted,” said Nolan.
“If you ask me, I would say that we'll never be able to fully understand and model cell function if we don't include spatial proteomics data.” — Emma Lundberg
Meanwhile keynote speaker Emma Lundberg, an associate professor of bioengineering at Stanford and co-director of the Human Protein Atlas, outlined how her team has mapped where proteins are located in cells — a process known as "spatial proteomics." Interestingly, over half of proteins can be found in more than one part of the cell, which changes how they function.
“If you ask me, I would say that we'll never be able to fully understand and model cell function if we don't include spatial proteomics data,” said Lundberg.
Panelists also discussed next steps for engineered tissues and artificial organs in disease study and regenerative medicine. Sangeeta Bhatia, a professor of health sciences and of electrical engineering and computer science at MIT’s Koch Institute, said that researchers have been able to engineer artificial tissues and organs that have little structure, like the skin and cartilage, for decades. Now, they're moving onto endocrine tissue, like the pancreas and liver. “Then you start to think about the tissues whose function is dependent on architecture, like the kidney, the lung — that's the next frontier, and I think we are not quite there yet,” she said.
One challenge brought up by Paola Arlotta, a professor of stem cell and regenerative biology at Harvard University, is how to factor genetics into tissue and organ models. One way to do this is to see how cells from different individuals respond to the same kinds of disturbances. If researchers don’t take genetic variability into account, “we’re ignoring a fundamental component of what human disease is,” she said.
Computational and technological challenges
Keynote speaker Dana Pe’er, chair of the Computational and Systems Biology Program at the Sloan Kettering Institute, outlined computational limitations that need to be addressed to answer pressing biological questions. For example, as researchers move from profiling a small section of a tissue to mapping a whole tissue or organ in different samples, they need to be able to map different tissue sections to each other.
“We’re still largely trying to figure out how to process this data, which is hampering our ability to interpret and powerfully utilize the data,” Pe’er said.
Given that there’s not yet a spatial profiling technology that can provide both high resolution and high content information on features like proteins, researchers will often need to combine a spatial profiling method with single cell data.
Barbara Engelhardt, an associate professor of computer science at Princeton University, said taking multiple images from the same type of tissue and aligning them can help researchers better understand cell type variability.
At the end of the second panel, Anthony Philippakis, co-director of the Eric and Wendy Schmidt Center and chief data officer of the Broad Institute, asked panelists whether they had any “recipes for success” to foster collaborations between the two fields.
Bhatia emphasized the importance of having researchers, or research teams, who are “bilingual” — that is, able to understand both experimental and computational biology. "It doesn't work well if you're just the recipient of data and you don't understand the context." Bhatia said. "We have to create these teams where we can really speak both languages."
Starting the conversations needed to build this bilingual proficiency was precisely the goal of the workshop.
Wengong Jin planned to research language processing for his computer science PhD. But when Jin learned about research on machine-learning for drug discovery at the MIT Computer Science and Artificial Intelligence Laboratory, he told his advisor, Regina Barzilay, that he’d had a change of heart.
“She thought I was jet lagged, because I’d just come over from China and I was proposing a really big switch,” he said.
Jin, now a fellow at the Eric and Wendy Schmidt Center, stayed the course. Six years later, he and a team of researchers have come up with a new kind of model to automatically design antibodies — holding huge potential for immunotherapy.
Meanwhile, another Eric and Wendy Schmidt Center Fellow, PhD candidate Adit Radhakrishnan, recently developed a simple yet powerful method for virtually screening new drug candidates. That framework appears in a study published this April in Proceedings of the National Academy of Sciences.
“A number of research institutes have started using machine learning to answer key questions in biology. But at the Eric and Wendy Schmidt Center, as Jin’s and Radhakrishnan’s research shows, our goal is to also go in the other direction, by using biomedical problems to drive advances in machine-learning,” said Caroline Uhler, co-Director of the Eric and Wendy Schmidt Center, a core member of the Broad Institute, and professor in the Department of Electrical Engineering and Computer Science and the Institute for Data, Systems and Society at MIT.
Game-changer for antibody design
Discovering drugs has traditionally been a labor-intensive process, with researchers toiling away for years to test millions of molecules only to come up with a handful of candidates. Now, researchers like Jin and Radhakrishnan are working to automate that process.
“The idea is that we don't need experts to get a cup of coffee and then work all night trying to figure out a new molecule, but rather, to let the machine do the heavy lifting,” Jin said.
Wengong Jin
During his PhD, Jin was part of a research team that developed a machine-learning algorithm to speed up antibiotic discovery. The researchers found a new antibiotic that was effective against bacteria that are resistant to multiple drugs. In this instance, the team provided the model with roughly a million possible compounds to sort through.
That left Jin and other researchers wondering: Could they use artificial intelligence to design molecules from scratch?
The answer was yes. Jin and other researchers developed a generative model that designed antibodies — Y-shaped proteins that bind to viruses, bacteria, and other pathogens, activating our bodies’ immune response — that could neutralize the SARS-CoV-2 virus. Their findings were published earlier this year in a paper at the International Conference on Learning Representations.
"The new model can propose in a couple of seconds an antibody that has a high likelihood of working — totally changing the game,” said Jin.
While researchers had worked on generative models for antibody discovery before, those models could only come up with a protein’s amino acid sequence — not its shape. In contrast, the new model, which represents the antibody as a graph, simultaneously designs both the sequence and structure of its binding region. “Whether or not the antibody is the right shape to bind to a virus or other pathogen is crucial to its success,” said Jin.
“The new model can propose in a couple of seconds an antibody that has a high likelihood of working — totally changing the game." — Wengong Jin
"While human experts have methods to generate neutralizing antibodies, it takes time and effort. The task becomes even more challenging when additional properties need to be enforced. As our understanding of disease biology and immune system deepens, the number of such desired characteristics will continue to grow. Computational methods for antibody design are particularly useful to address this challenge,” said Regina Barzilay, the AI faculty lead for the MIT Jameel Clinic for Machine Learning in Health.
And, because so many types of data are structured as networks, the model also represents an advance in the field of machine learning. “It’s an example of how biology proposed a new problem for machine learning to solve,” said Jin.
An old machine-learning method repurposed for virtual drug screening
Adit Radhakrishnan's father had pursued a mathematics education in India prior to immigrating to the U.S. He instilled in his son a love of math, which led the younger Radhakrishnan to pursue a PhD of his own in electrical engineering and computer science at MIT.
Radhakrishnan researches the fundamentals of deep learning — a kind of artificial intelligence modeled after the human brain that processes unstructured data. Understanding why deep learning is successful, and using that knowledge to build novel models for the healthcare and genomic space, underpins much of Radhakrishnan’s research as an Eric and Wendy Schmidt Center fellow.
Adit Radhakrishnan
Over the past few years, deep learning has become widely adopted in biological applications, with researchers increasingly turning to it to screen potential new drugs. In order to perform well on such tasks, researchers use very large deep learning models that often require significant computing power. Moreover, the complexity of this approach makes it hard for scientists to understand why these models make a given prediction, shedding little light on why a proposed drug could work.
To get around the complexities of deep learning, Radhakrishnan and other researchers, including Uhler and Mikhail Belkin, a professor at the Halıcıoğlu Data Science Institute at the University of California, San Diego, turned to an older class of machine learning models: kernel methods. Prior to the recent wave of deep learning, kernel methods were a prominent and computationally simple approach for machine learning tasks. These models have recently become popular again since they can serve as a proxy for using very large deep learning models with much less computational burden.
The team came up with a simple yet highly adaptable kernel framework that was able to predict the effect that a drug has on gene expression, a measure of how cells change in response to a drug. “In contrast to the expertise needed to train large deep learning models to solve a particular problem, it takes about three lines of code to train the kernel method to do the same task,” said Radhakrishnan.
The framework has uses beyond biology; the researchers demonstrated, for example, that it could be used by video streaming providers to predict how a viewer would rank a particular movie they hadn’t yet seen. And the framework allows researchers to gain insights into how more complex deep learning models function.
According to Radhakrishnan, who is not trained as a biologist, the best part of being a fellow at the Eric and Wendy Schmidt Center is that the center puts machine learning experts and biologists in constant conversation with each other.
“You don’t just have computational researchers running their methods on a biology dataset without a biologist in the mix. You can get continuous feedback on: Is this actually useful?” said Radhakrishnan. “So it gives you a much more guided focus on what biological problems are important and what computational methods are missing.”














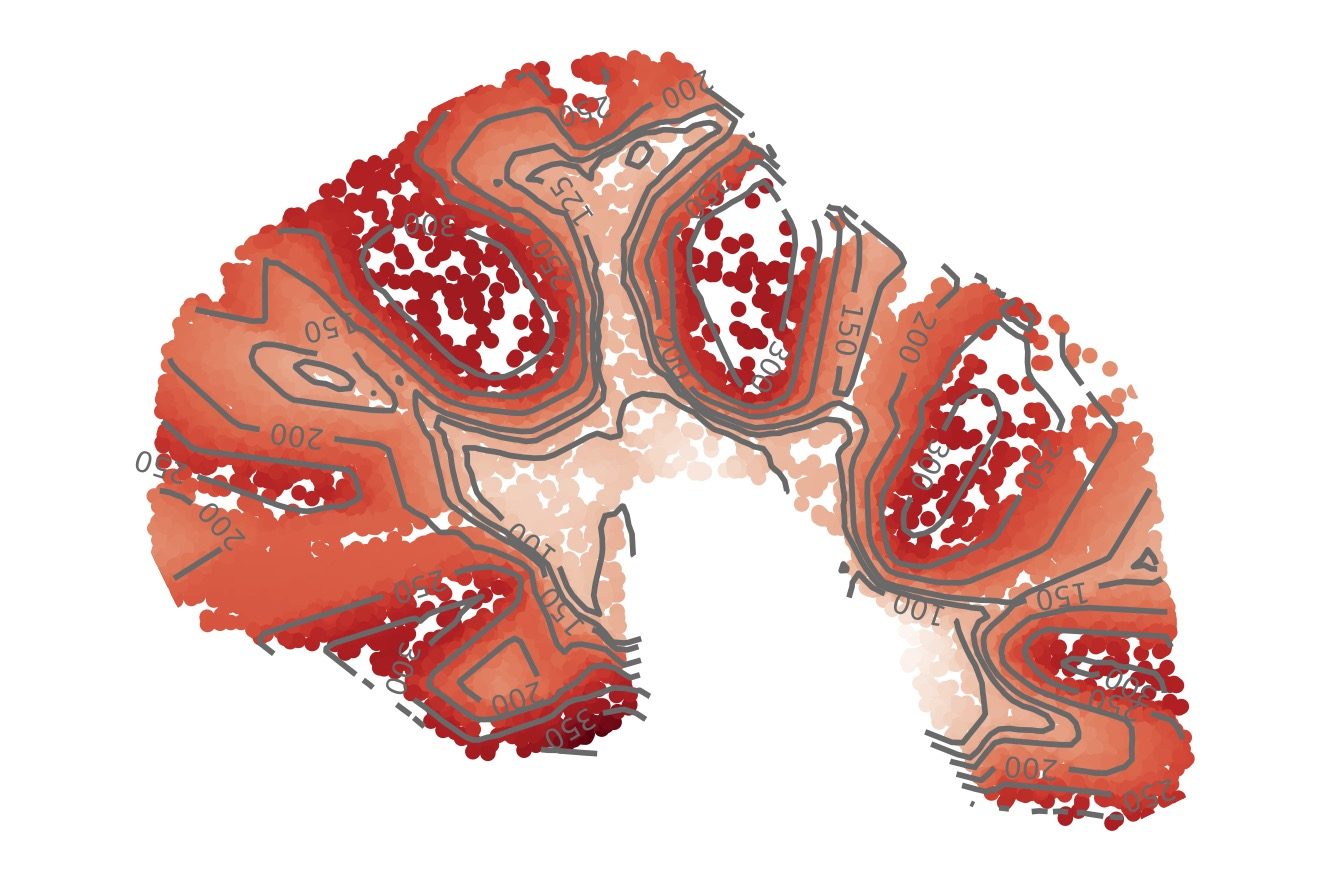





.png)









.png)
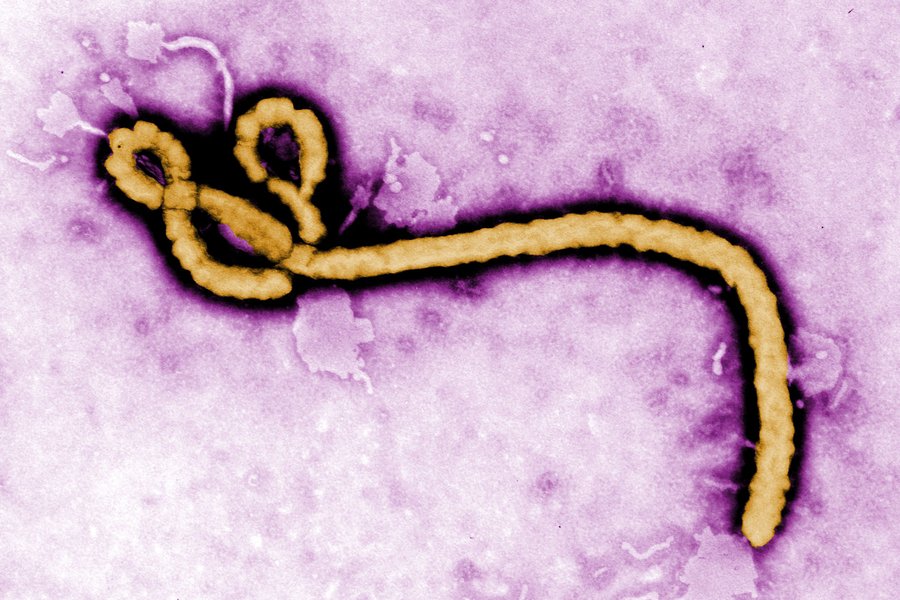















































%20copy.jpg)










.png)








.jpg)



.jpg)


.jpg)


-fotor-2024013094636.png)


































